

Sporadic Creutzfeldt-Jakob Disease in a Very Young Person
- PMID: 34497065
- PMCID: PMC8605619
- DOI: 10.1212/WNL.0000000000012737
Sporadic Creutzfeldt-Jakob Disease in a Very Young Person
Abstract
Background and objectives: Sporadic Creutzfeldt-Jakob disease (sCJD) is the most common form of human prion disease and typically occurs in middle to late life. sCJD in early adulthood is extremely uncommon. The purpose of this report is to raise awareness of cases of sCJD in young patients that are not associated with a genetic mutation or acquired prion disease risk factors.
Methods: We describe the clinical presentation, diagnostic workup, and postmortem examination of a 22-year-old man with sCJD.
Results: The patient presented with a rapidly progressive neurocognitive disorder consisting of early and prominent psychiatric symptoms. CSF real-time quaking-induced conversion (RT-QuIC) was indeterminate, and brain MRI was suggestive of prion disease. Neuropathologic examination and the absence of a genetic mutation and acquired prion disease risk factors resulted in a final diagnosis of sCJD.
Conclusion: Although extremely rare, sCJD can occur in young people and should be considered in the setting of rapidly progressive neuropsychiatric conditions. Postmortem examination is required to diagnose the type of prion disease and remains important to surveil for known and potentially novel acquired prion diseases.
© 2021 American Academy of Neurology.
Figures
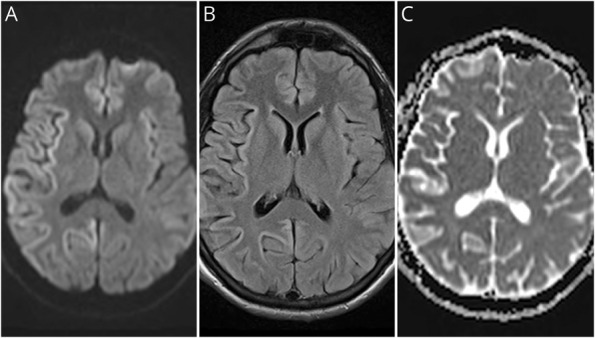
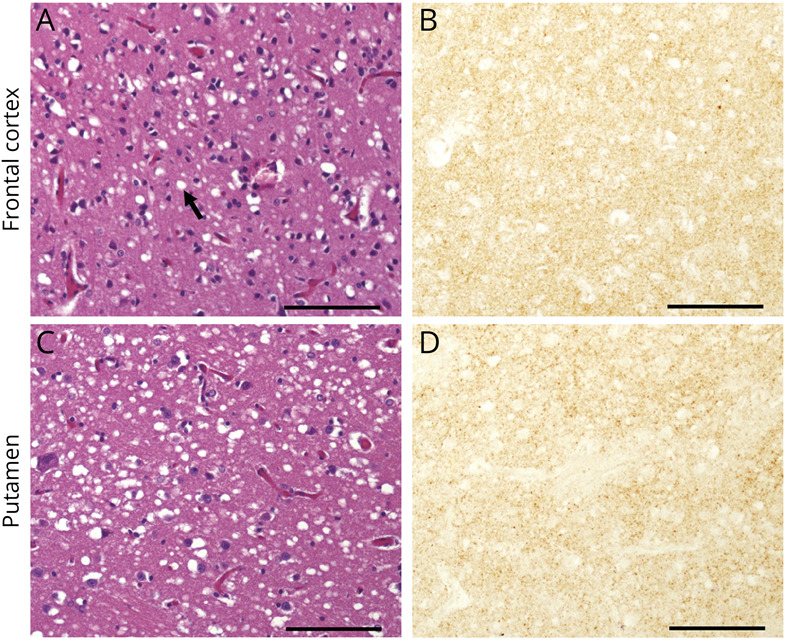
Comment in
-
The Intractable Puzzle of Sporadic Creutzfeldt-Jakob Disease in Very Young People.Neurology. 2021 Oct 26;97(17):801-802. doi: 10.1212/WNL.0000000000012739. Epub 2021 Sep 8. Neurology. 2021. PMID: 34497066 No abstract available.
Comment on
-
The Intractable Puzzle of Sporadic Creutzfeldt-Jakob Disease in Very Young People.Neurology. 2021 Oct 26;97(17):801-802. doi: 10.1212/WNL.0000000000012739. Epub 2021 Sep 8. Neurology. 2021. PMID: 34497066 No abstract available.
References
-
- Maddox RA, Person MK, Blevins JE, et al. Prion disease incidence in the United States, 2003-2015. Neurology. 2020;94(2):e153-e157. - PubMed
-
- Thompson AGB, Lowe J, Fox Z, et al. The Medical Research Council prion disease rating scale: a new outcome measure for prion disease therapeutic trials developed and validated using systematic observational studies. Brain. 2013;136(pt 4):1116-1127. - PubMed
-
- Rhoads DD, Wrona A, Foutz A, et al. Diagnosis of prion diseases by RT-QuIC results in improved surveillance. Neurology. 2020;95(8):e1017-e1026. - PubMed
-
- Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46(2):224-233. - PubMed
-
- Meissner B, Westner IM, Kallenberg K, et al. Sporadic Creutzfeldt-Jakob disease: clinical and diagnostic characteristics of the rare VV1 type. Neurology. 2005;65(10):1544-1550. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical