

Dual RNASeq Reveals NTHi-Macrophage Transcriptomic Changes During Intracellular Persistence
- PMID: 34497778
- PMCID: PMC8419319
- DOI: 10.3389/fcimb.2021.723481
Dual RNASeq Reveals NTHi-Macrophage Transcriptomic Changes During Intracellular Persistence
Abstract
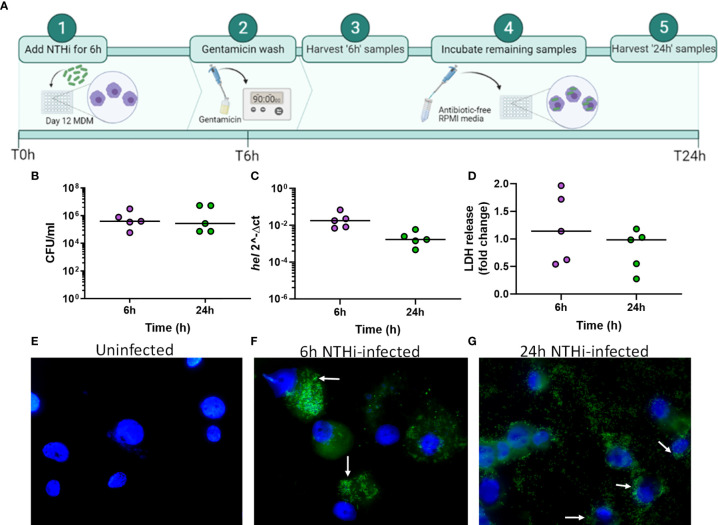
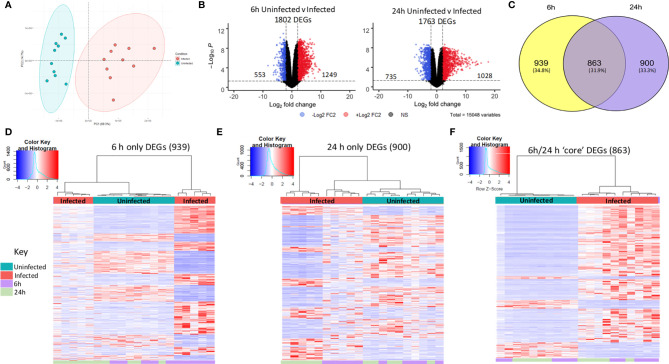
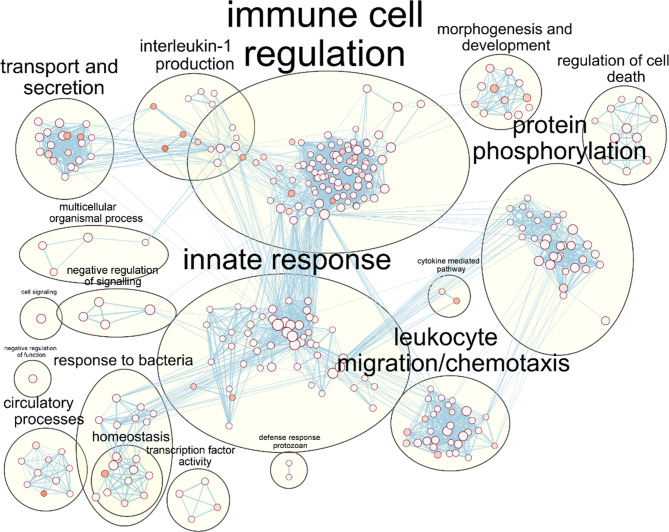
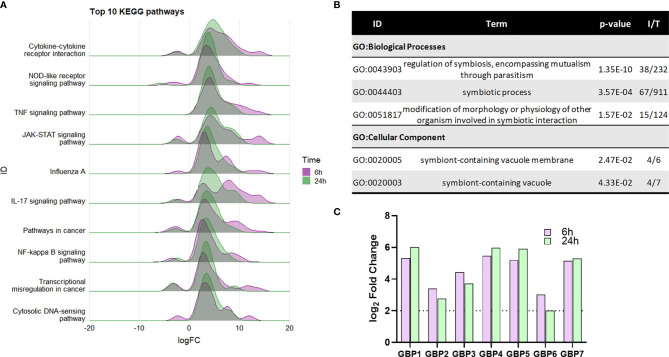
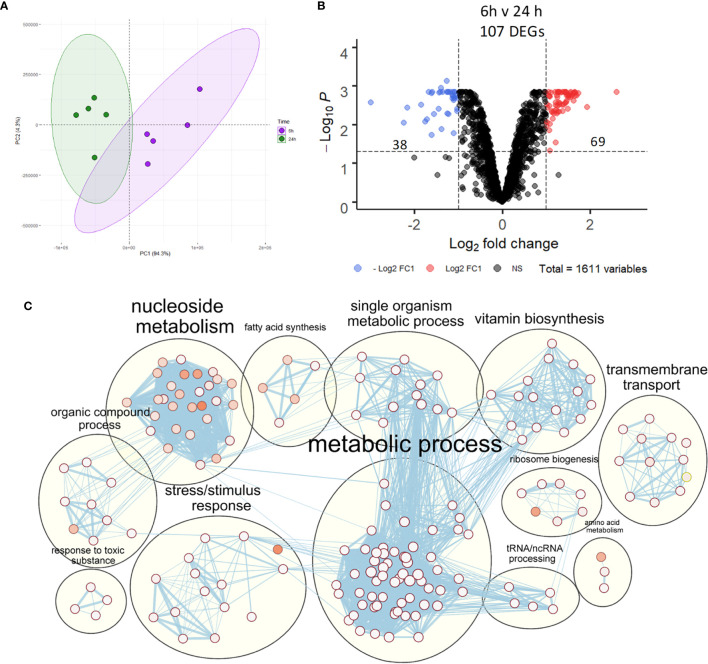
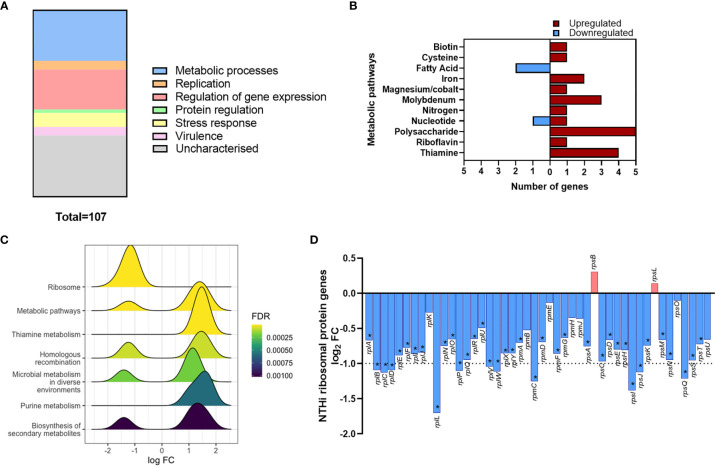
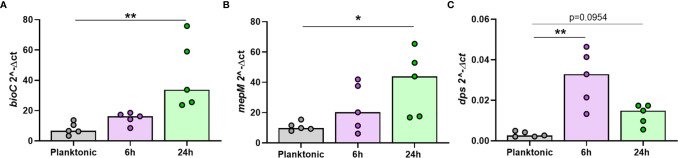
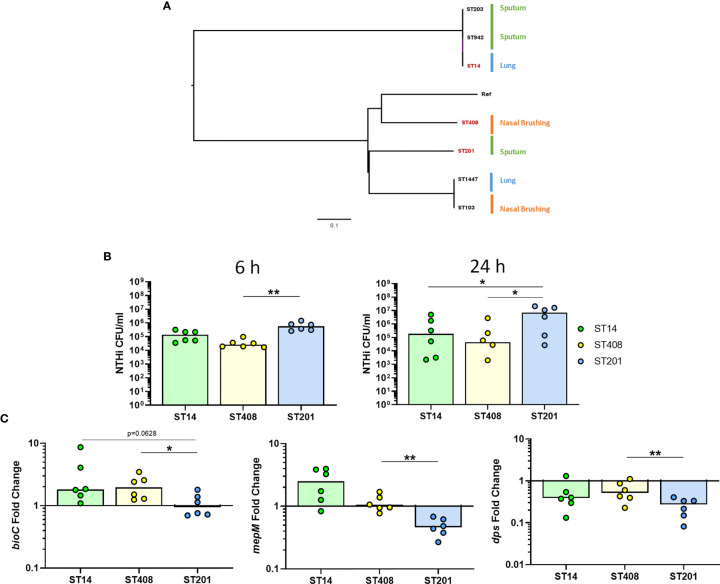
Nontypeable Haemophilus influenzae (NTHi) is a pathobiont which chronically colonises the airway of individuals with chronic respiratory disease and is associated with poor clinical outcomes. It is unclear how NTHi persists in the airway, however accumulating evidence suggests that NTHi can invade and persist within macrophages. To better understand the mechanisms of NTHi persistence within macrophages, we developed an in vitro model of NTHi intracellular persistence using human monocyte-derived macrophages (MDM). Dual RNA Sequencing was used to assess MDM and NTHi transcriptomic regulation occurring simultaneously during NTHi persistence. Analysis of the macrophage response to NTHi identified temporally regulated transcriptomic profiles, with a specific 'core' profile displaying conserved expression of genes across time points. Gene list enrichment analysis identified enrichment of immune responses in the core gene set, with KEGG pathway analysis revealing specific enrichment of intracellular immune response pathways. NTHi persistence was facilitated by modulation of bacterial metabolic, stress response and ribosome pathways. Levels of NTHi genes bioC, mepM and dps were differentially expressed by intracellular NTHi compared to planktonic NTHi, indicating that the transcriptomic adaption was distinct between the two different NTHi lifestyles. Overall, this study provides crucial insights into the transcriptomic adaptations facilitating NTHi persistence within macrophages. Targeting these reported pathways with novel therapeutics to reduce NTHi burden in the airway could be an effective treatment strategy given the current antimicrobial resistance crisis and lack of NTHi vaccines.
Keywords: NTHi; dual RNAseq; host-pathogen interactions; intracellular persistence; macrophage.
Copyright © 2021 Ackland, Heinson, Cleary, Christodoulides, Wilkinson and Staples.
Conflict of interest statement
DC reports that he was a post-doctoral researcher on projects funded by Pfizer and GSK between April 2014 and October 2017. TW reports grants and personal fees from AstraZeneca, personal fees and other from MMH, grants and personal fees from GSK, personal fees from BI, and grants and personal fees from Synairgen, outside the submitted work. KS reports grants from AstraZeneca, outside the submitted work. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases