

Best practices for analyzing imputed genotypes from low-pass sequencing in dogs
- PMID: 34498136
- PMCID: PMC8913487
- DOI: 10.1007/s00335-021-09914-z
Best practices for analyzing imputed genotypes from low-pass sequencing in dogs
Abstract
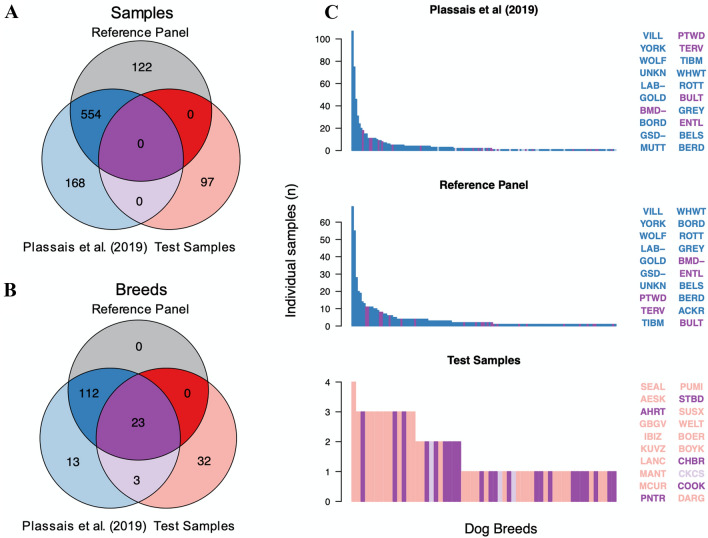
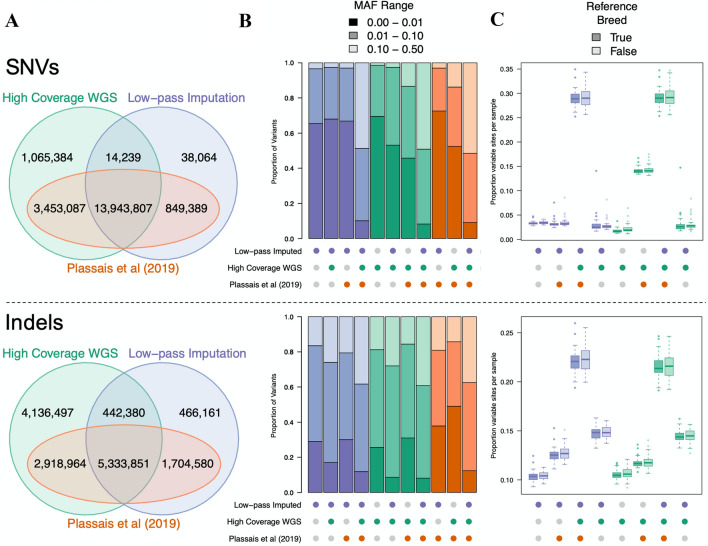
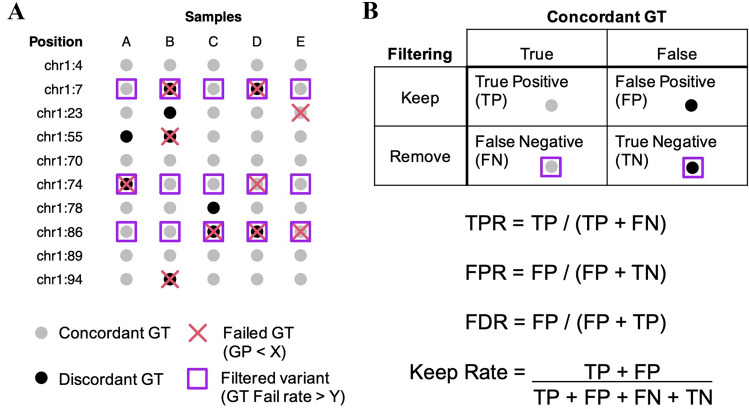
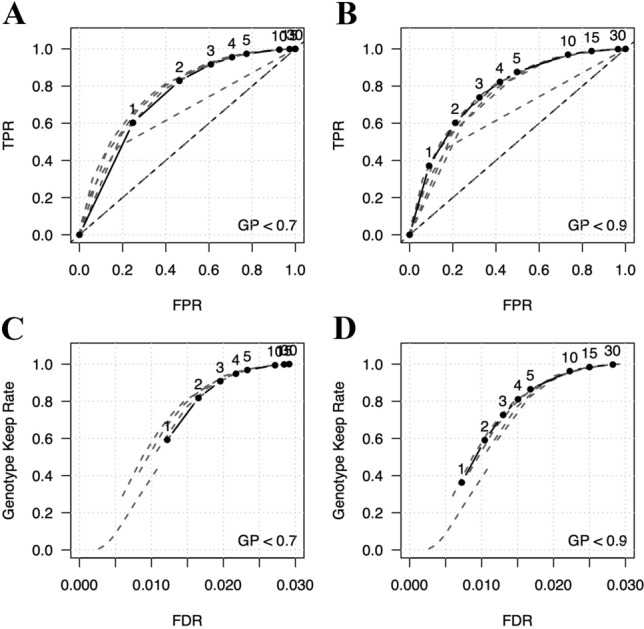
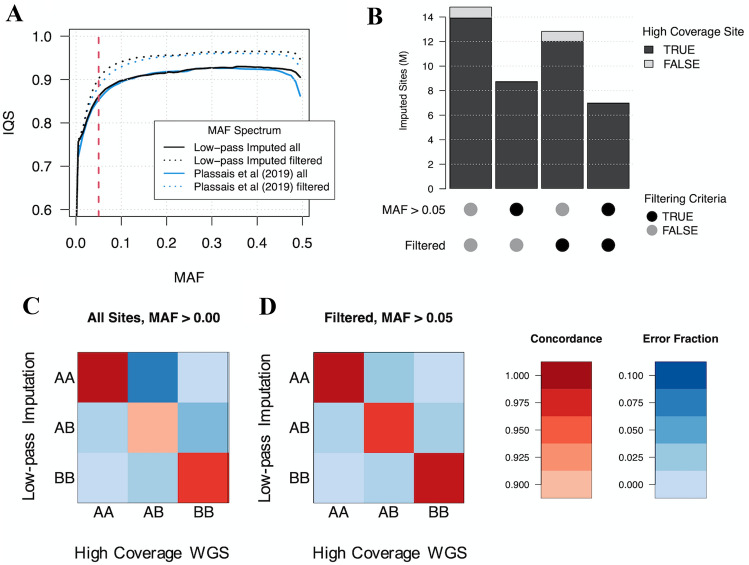
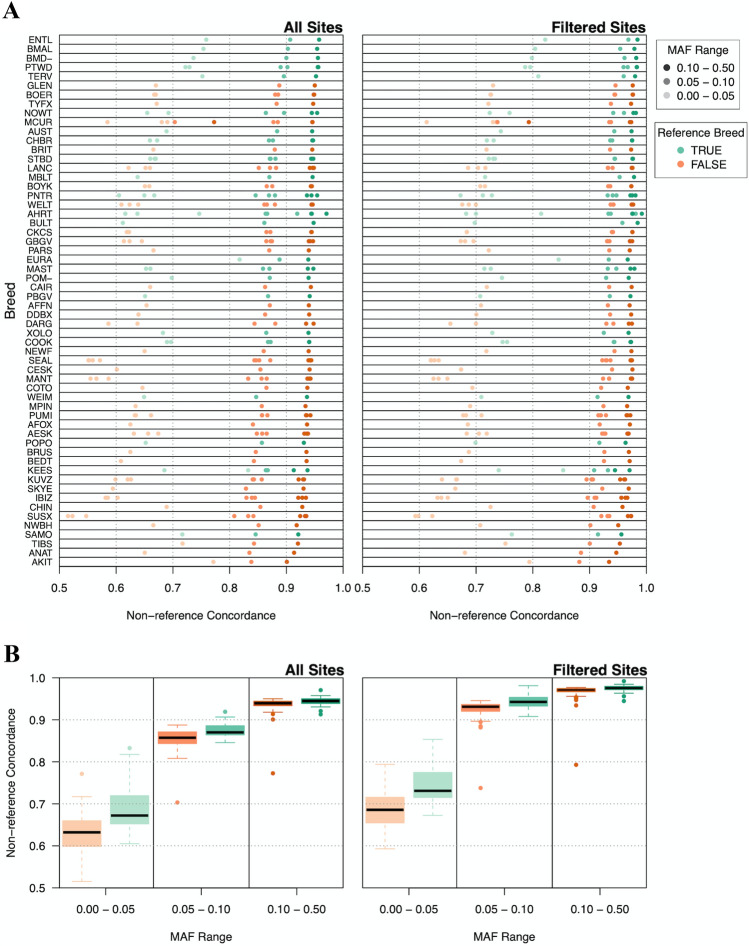
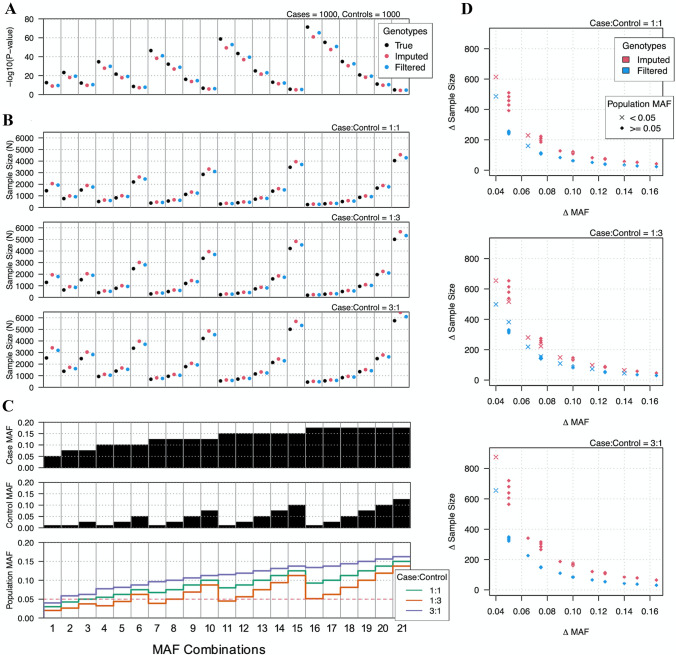
Although DNA array-based approaches for genome-wide association studies (GWAS) permit the collection of thousands of low-cost genotypes, it is often at the expense of resolution and completeness, as SNP chip technologies are ultimately limited by SNPs chosen during array development. An alternative low-cost approach is low-pass whole genome sequencing (WGS) followed by imputation. Rather than relying on high levels of genotype confidence at a set of select loci, low-pass WGS and imputation rely on the combined information from millions of randomly sampled low-confidence genotypes. To investigate low-pass WGS and imputation in the dog, we assessed accuracy and performance by downsampling 97 high-coverage (> 15×) WGS datasets from 51 different breeds to approximately 1× coverage, simulating low-pass WGS. Using a reference panel of 676 dogs from 91 breeds, genotypes were imputed from the downsampled data and compared to a truth set of genotypes generated from high-coverage WGS. Using our truth set, we optimized a variant quality filtering strategy that retained approximately 80% of 14 M imputed sites and lowered the imputation error rate from 3.0% to 1.5%. Seven million sites remained with a MAF > 5% and an average imputation quality score of 0.95. Finally, we simulated the impact of imputation errors on outcomes for case-control GWAS, where small effect sizes were most impacted and medium-to-large effect sizes were minorly impacted. These analyses provide best practice guidelines for study design and data post-processing of low-pass WGS-imputed genotypes in dogs.
© 2021. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
All authors declare no competing interests and that the presented work is original.
Figures
References
-
- Benjelloun B, Boyer F, Streeter I, Zamani W, Engelen S, Alberti A, Alberto FJ, BenBati M, Ibnelbachyr M, Chentouf M, Bechchari A, Rezaei HR, Naderi S, Stella A, Chikhi A, Clarke L, Kijas J, Flicek P, Taberlet P, Pompanon F. An evaluation of sequencing coverage and genotyping strategies to assess neutral and adaptive diversity. Mol Ecol Resour. 2019;19:1497–1515. - PMC - PubMed
-
- Boyko AR, Quignon P, Li L, Schoenebeck JJ, Degenhardt JD, Lohmueller KE, Zhao K, Brisbin A, Parker HG, vonHoldt BM, Cargill M, Auton A, Reynolds A, Elkahloun AG, Castelhano M, Mosher DS, Sutter NB, Johnson GS, Novembre J, Hubisz MJ, Siepel A, Wayne RK, Bustamante CD, Ostrander EA. A simple genetic architecture underlies morphological variation in dogs. PLoS Biol. 2010;8:e1000451. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
