

Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic
- PMID: 34499854
- PMCID: PMC8367733
- DOI: 10.1016/j.cell.2021.08.014
Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic
Abstract
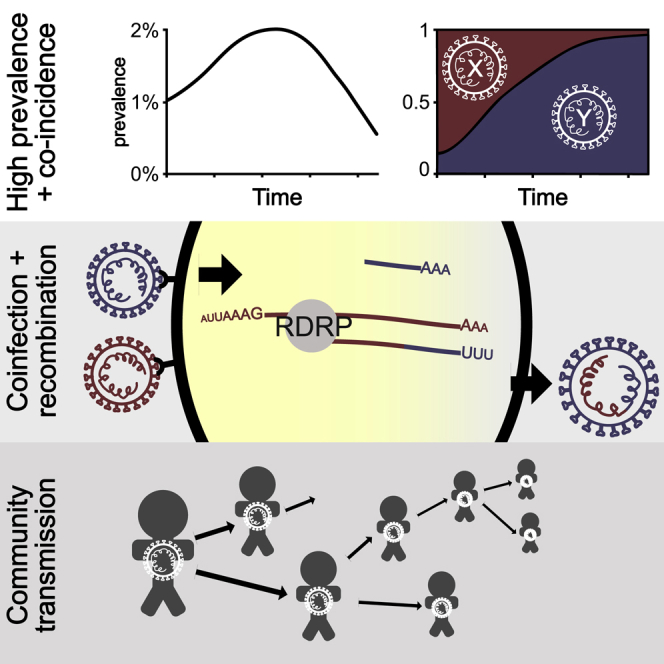
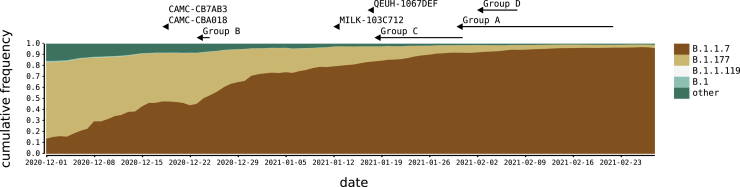
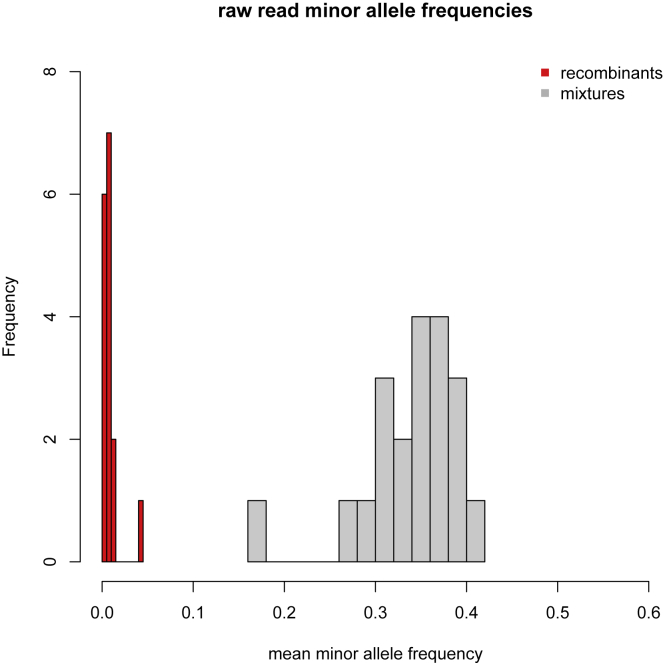
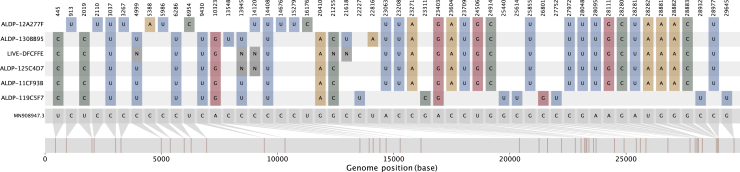
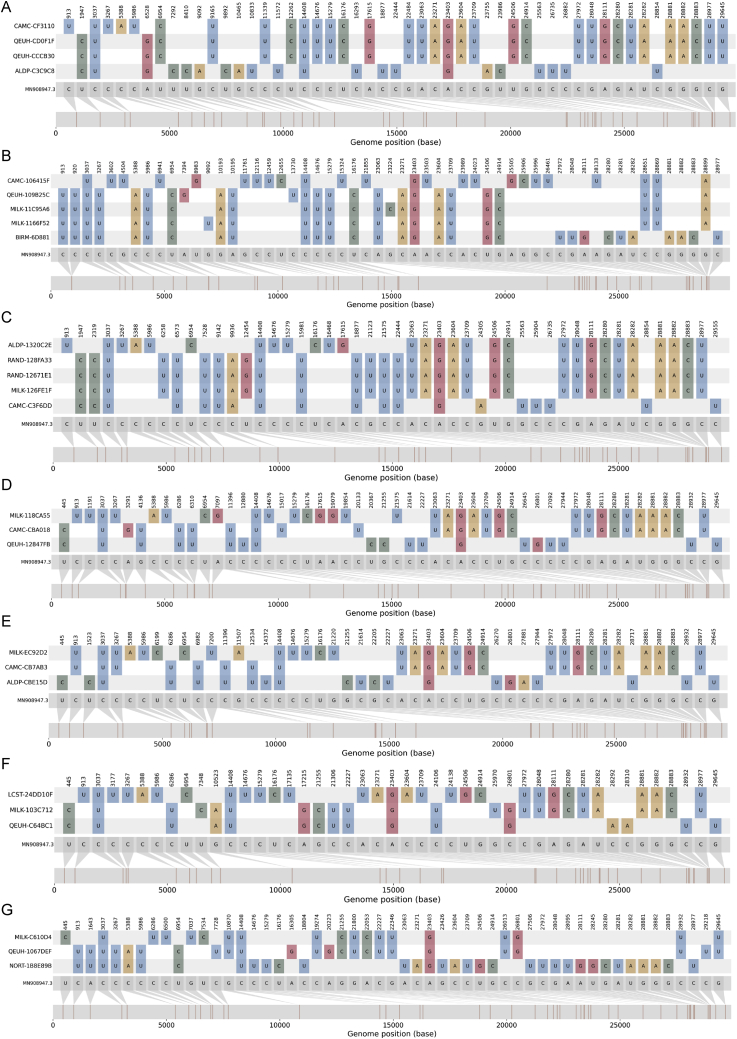
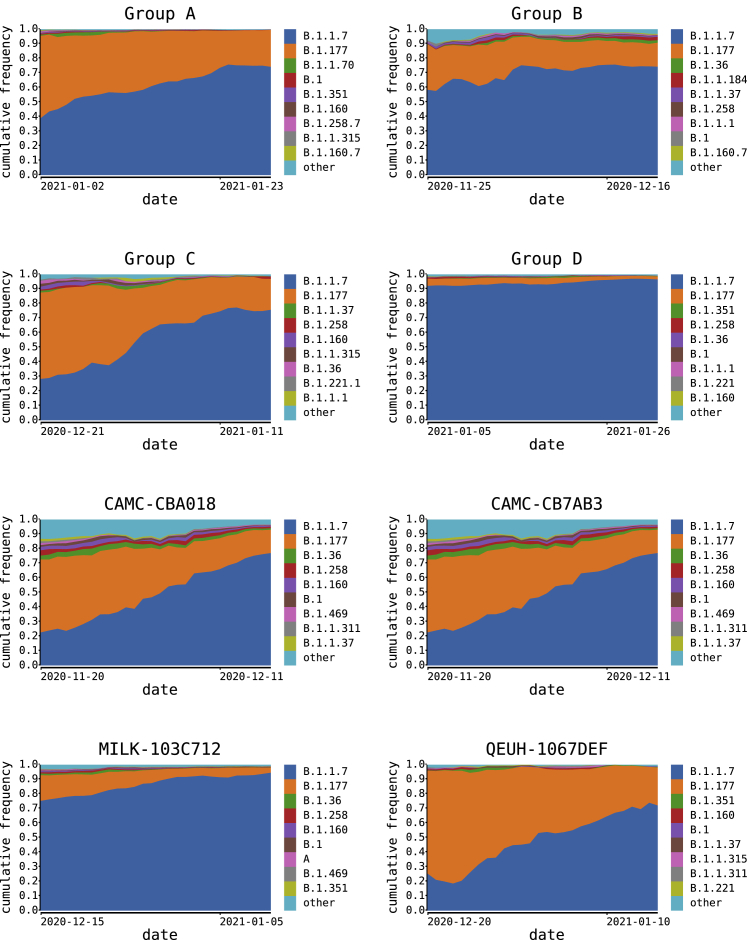
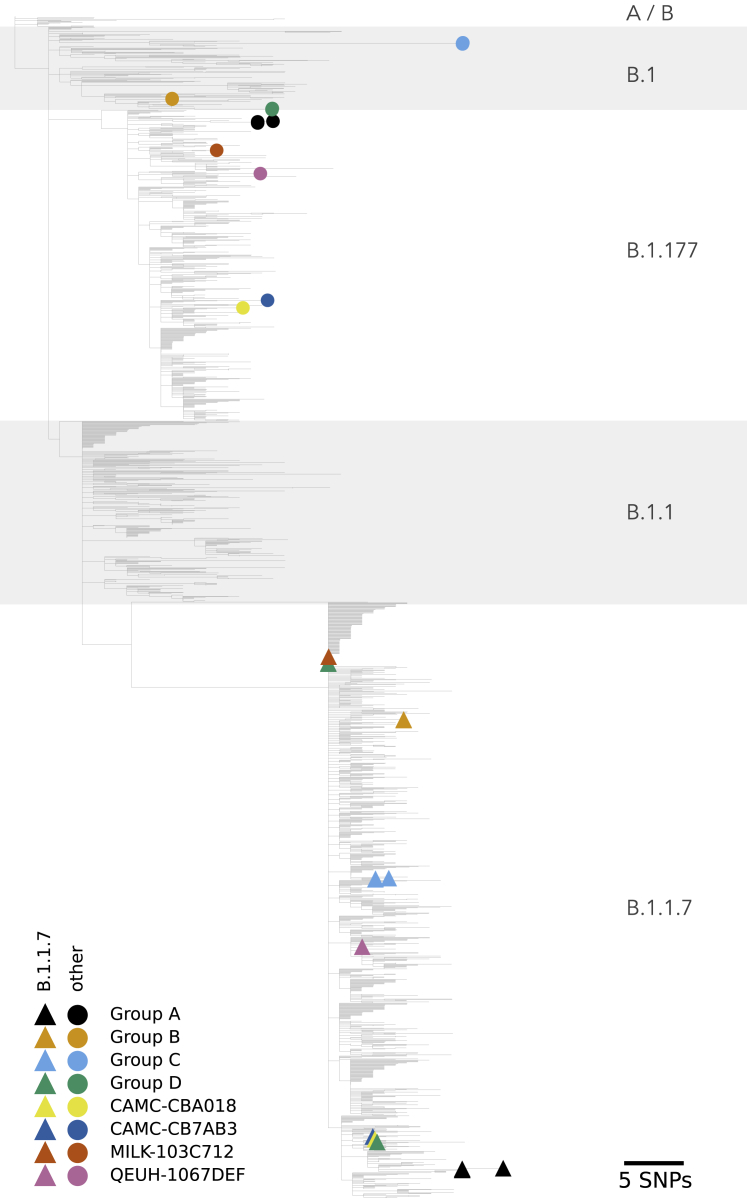
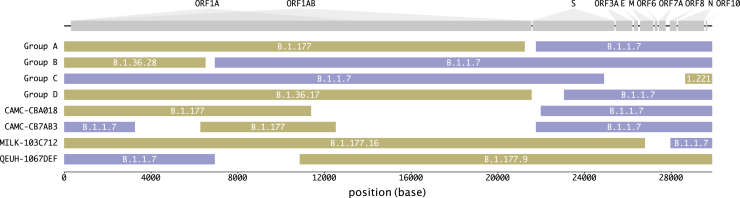
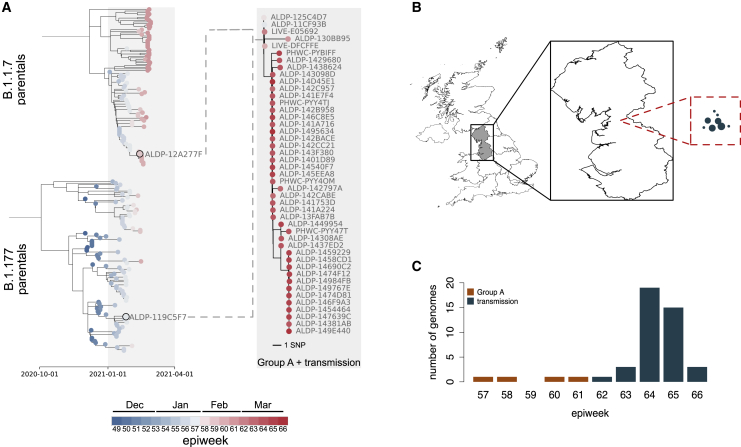
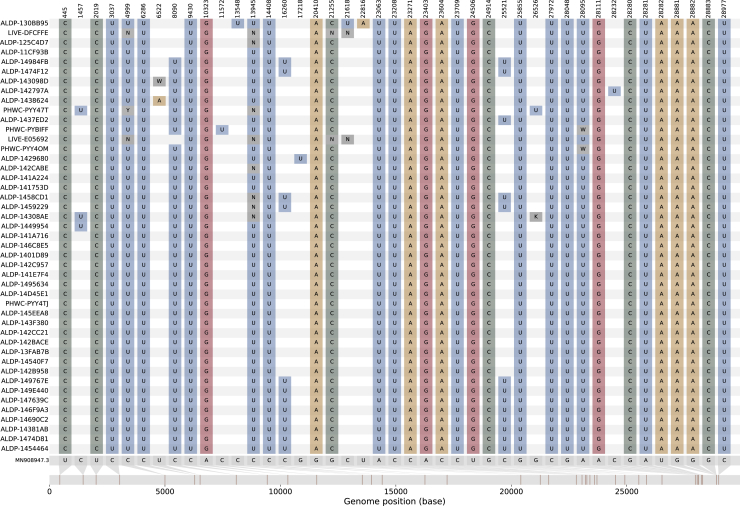
We present evidence for multiple independent origins of recombinant SARS-CoV-2 viruses sampled from late 2020 and early 2021 in the United Kingdom. Their genomes carry single-nucleotide polymorphisms and deletions that are characteristic of the B.1.1.7 variant of concern but lack the full complement of lineage-defining mutations. Instead, the remainder of their genomes share contiguous genetic variation with non-B.1.1.7 viruses circulating in the same geographic area at the same time as the recombinants. In four instances, there was evidence for onward transmission of a recombinant-origin virus, including one transmission cluster of 45 sequenced cases over the course of 2 months. The inferred genomic locations of recombination breakpoints suggest that every community-transmitted recombinant virus inherited its spike region from a B.1.1.7 parental virus, consistent with a transmission advantage for B.1.1.7's set of mutations.
Keywords: B.1.1.7; SARS-CoV-2; evolution; genomic epidemiology; genomics; recombination; variants.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Boni M.F., Lemey P., Jiang X., Lam T.T.-Y., Perry B.W., Castoe T.A., Rambaut A., Robertson D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020;5:1408–1417. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
