

Structural Biology-Based Exploration of Subtype-Selective Agonists for Peroxisome Proliferator-Activated Receptors
- PMID: 34502131
- PMCID: PMC8430769
- DOI: 10.3390/ijms22179223
Structural Biology-Based Exploration of Subtype-Selective Agonists for Peroxisome Proliferator-Activated Receptors
Abstract
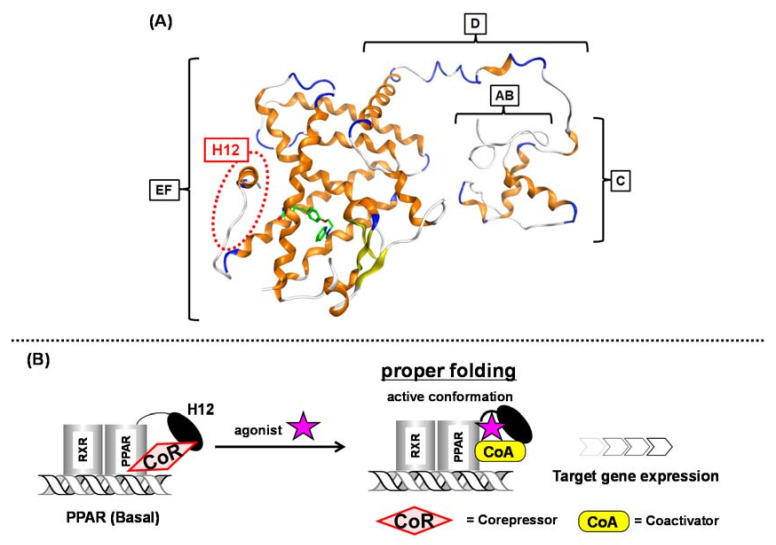
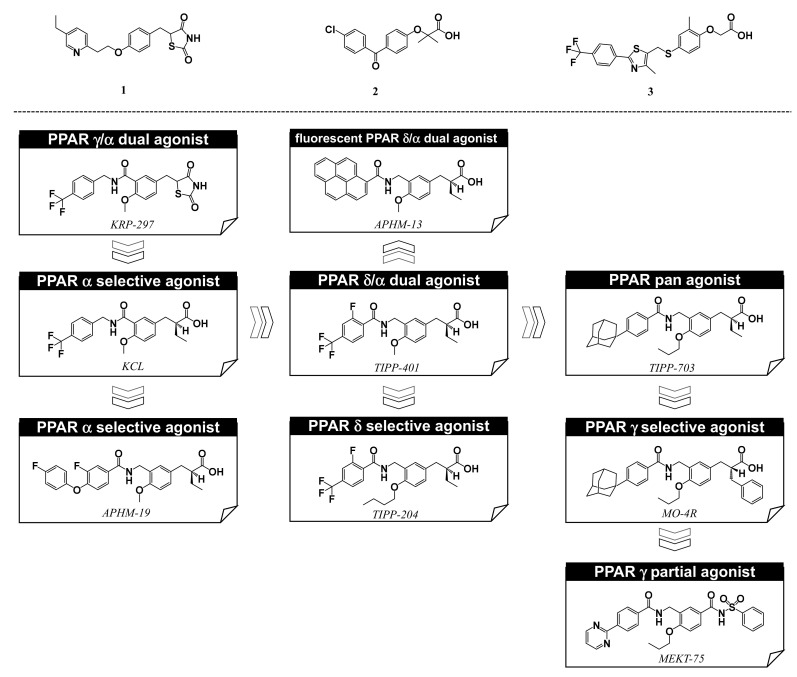
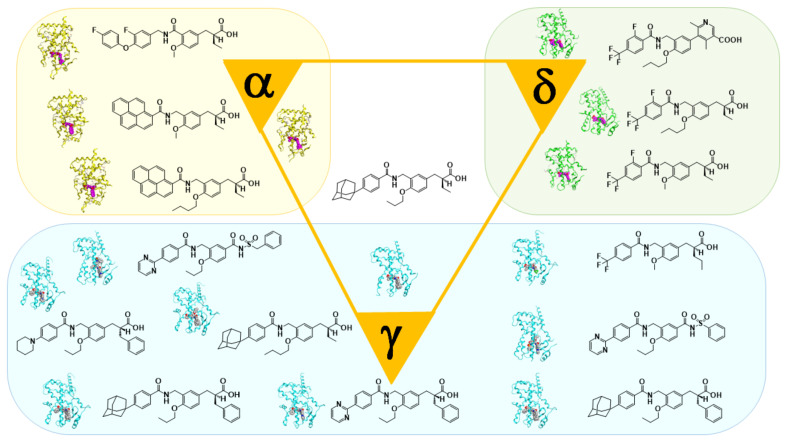
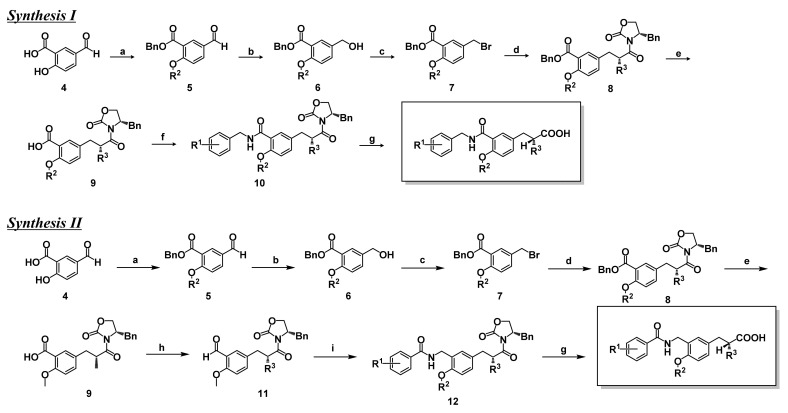
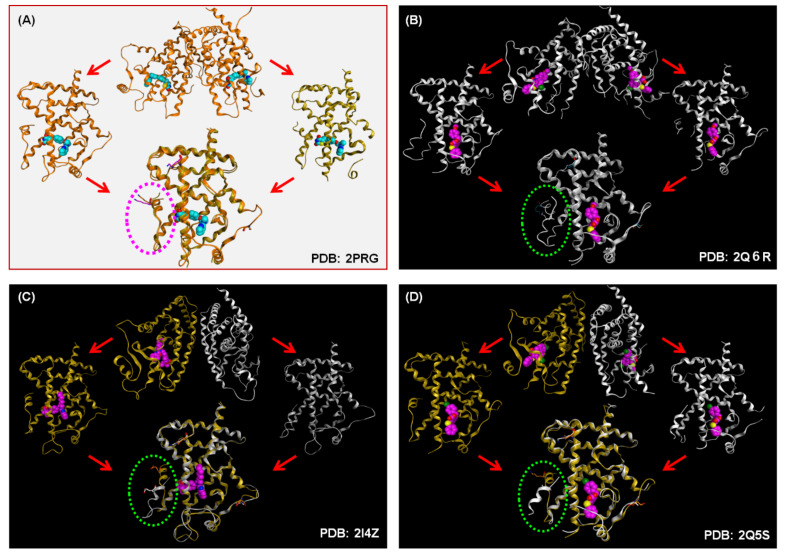
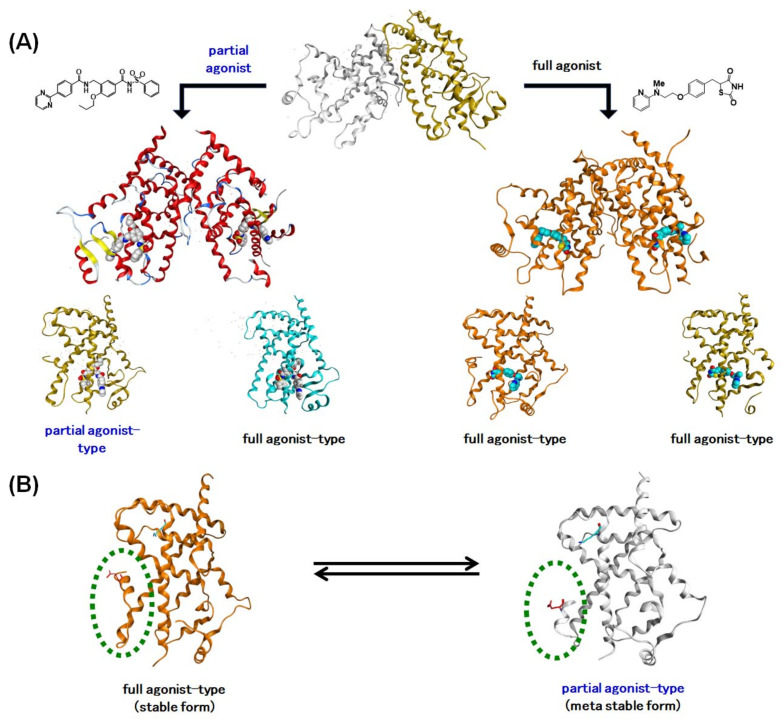
Progress in understanding peroxisome proliferator-activated receptor (PPAR) subtypes as nuclear receptors that have pleiotropic effects on biological responses has enabled the exploration of new subtype-selective PPAR ligands. Such ligands are useful chemical biology/pharmacological tools to investigate the functions of PPARs and are also candidate drugs for the treatment of PPAR-mediated diseases, such as metabolic syndrome, inflammation and cancer. This review summarizes our medicinal chemistry research of more than 20 years on the design, synthesis, and pharmacological evaluation of subtype-selective PPAR agonists, which has been based on two working hypotheses, the ligand superfamily concept and the helix 12 (H12) holding induction concept. X-ray crystallographic analyses of our agonists complexed with each PPAR subtype validate our working hypotheses.
Keywords: PPAR agonist; helix 12 holding induction concept; ligand superfamily concept; peroxisome proliferator-activated receptor; structural biology.
Conflict of interest statement
The author declares no conflict of interest.
Figures
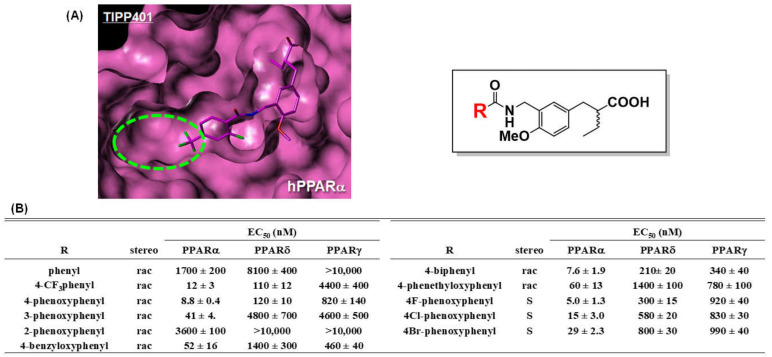
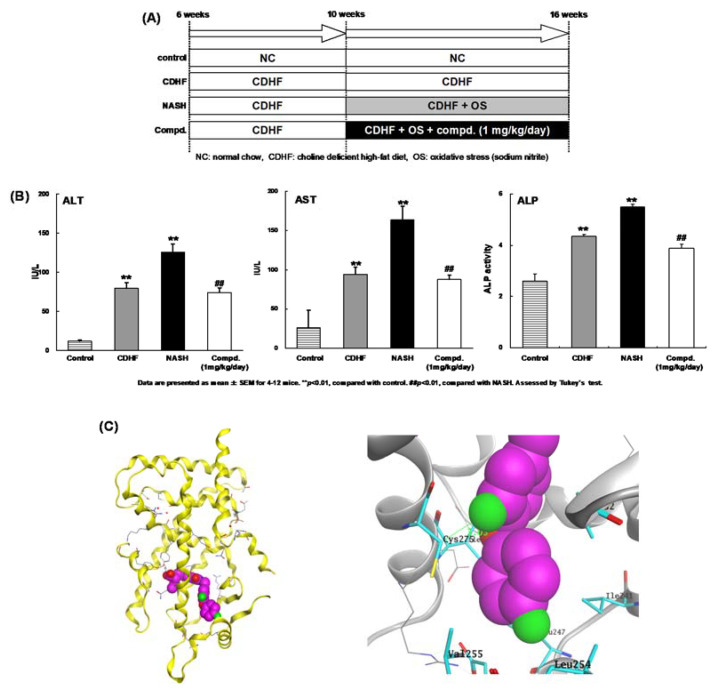
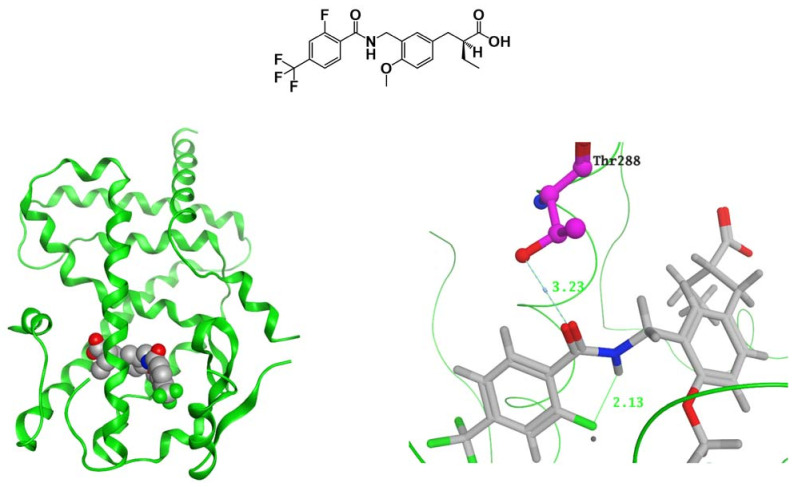
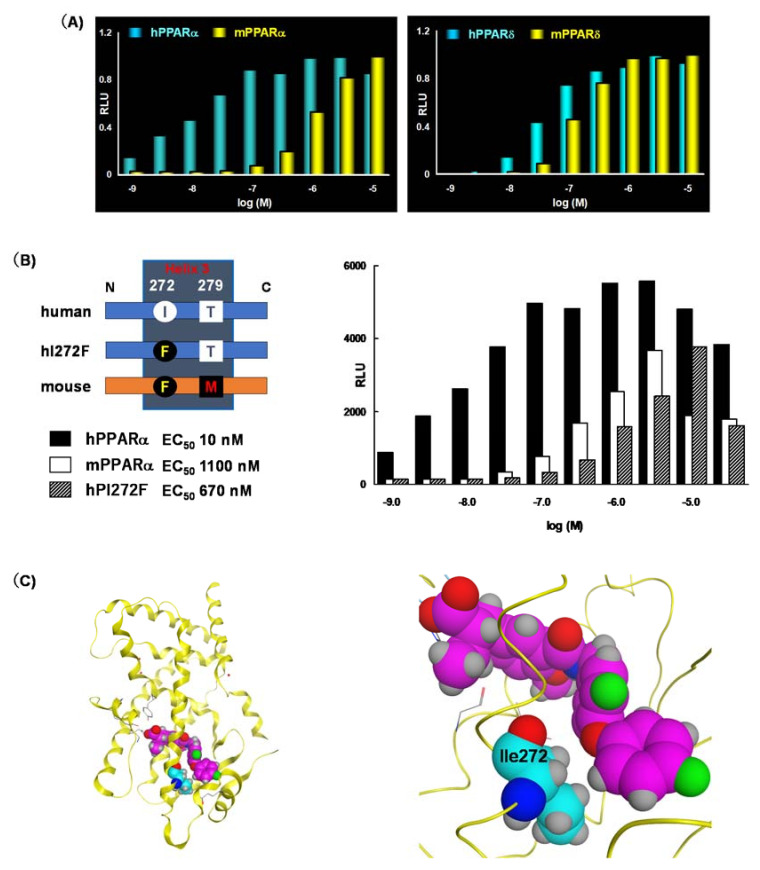

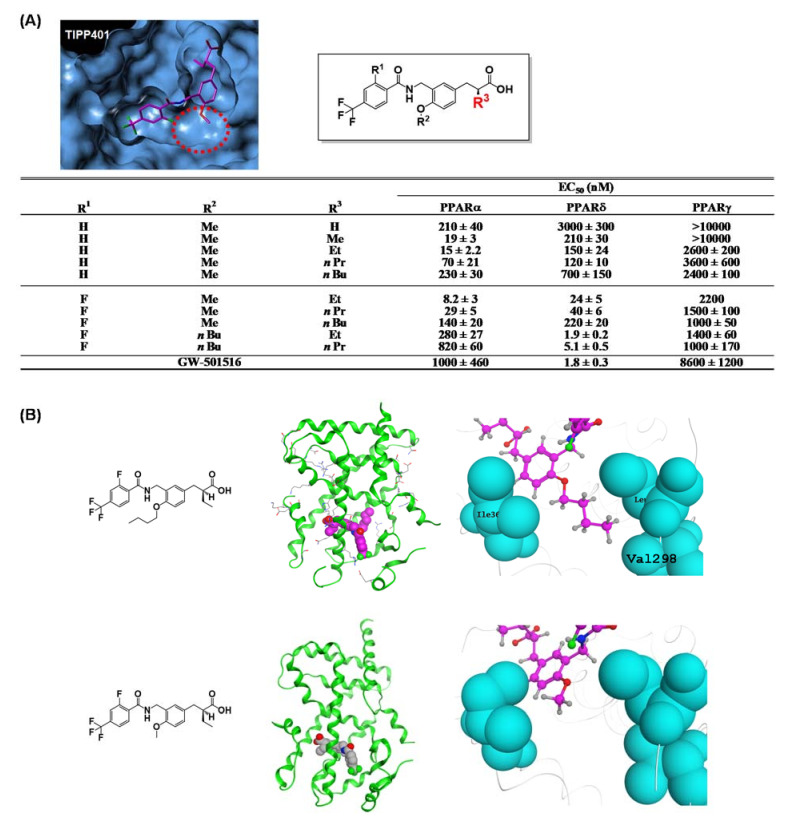
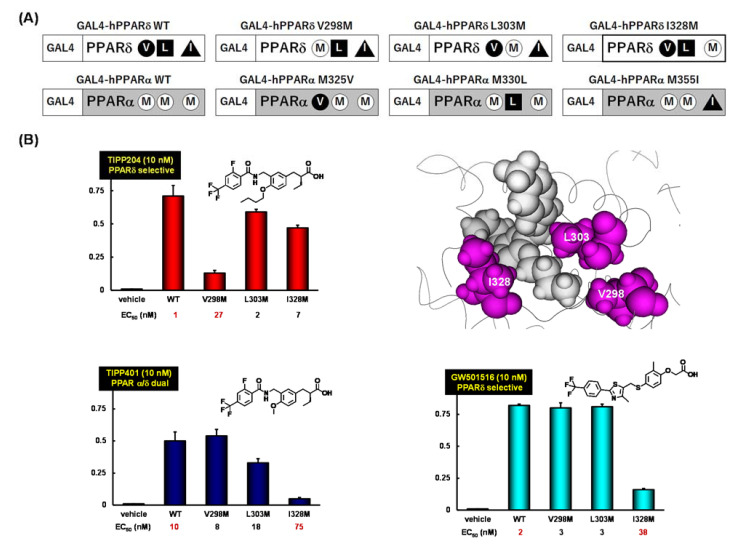
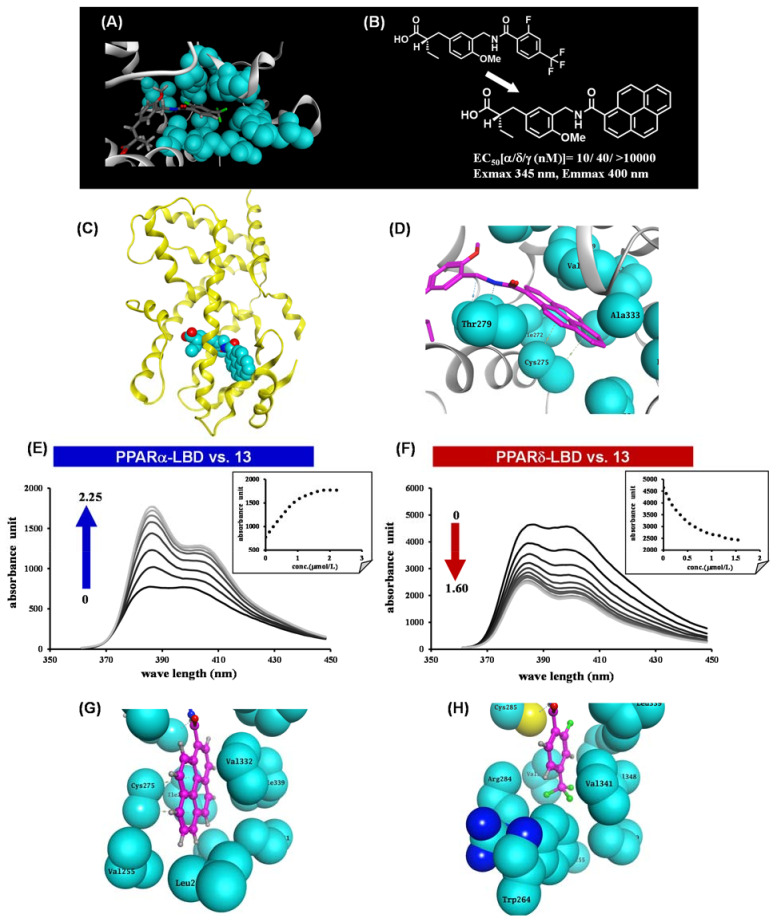
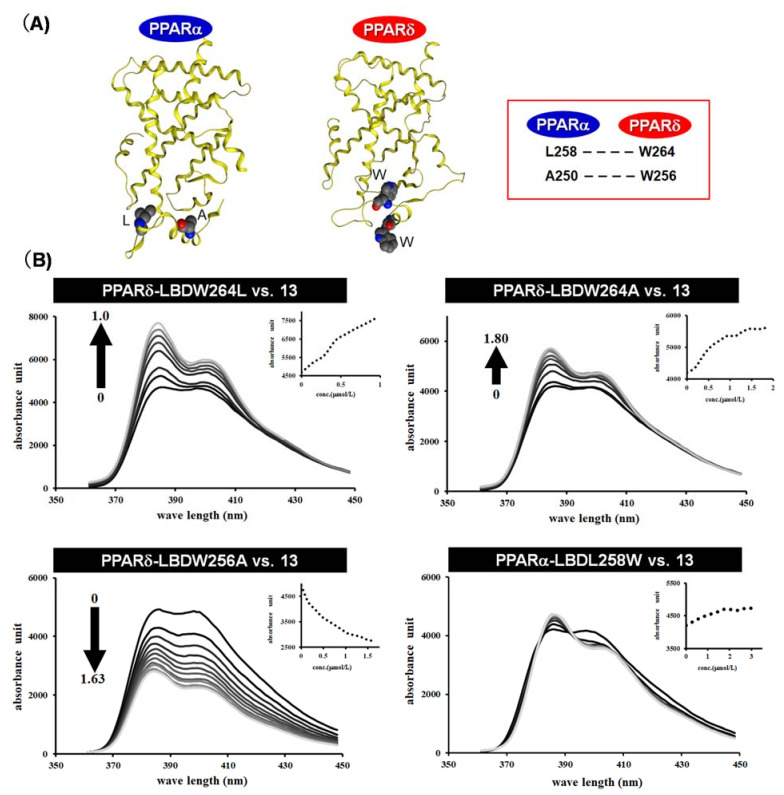
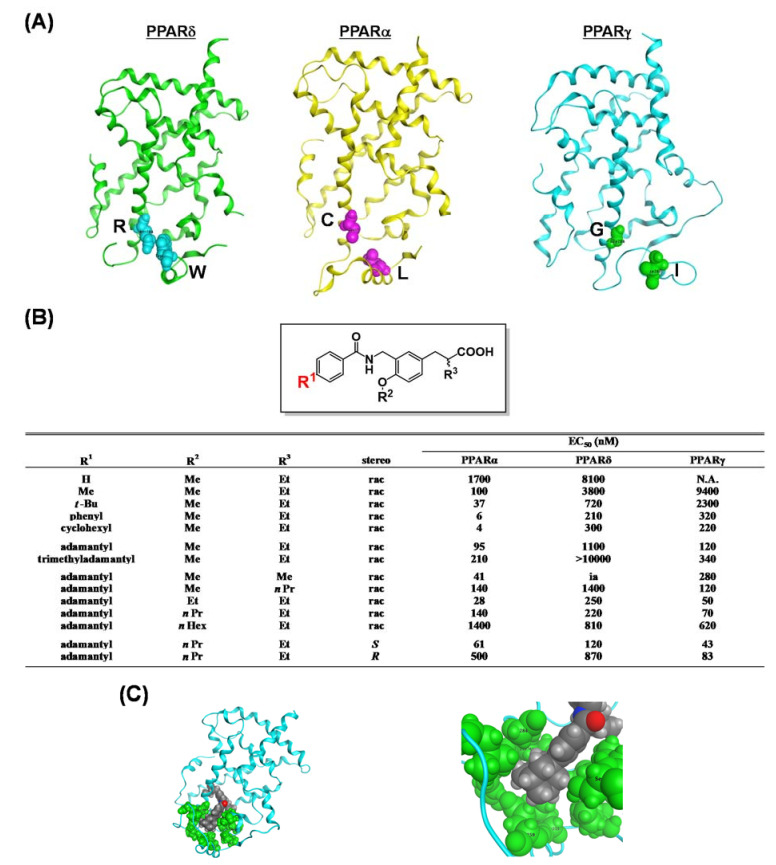
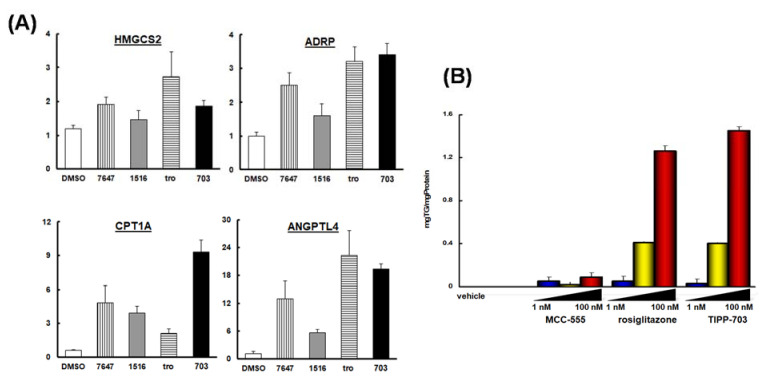
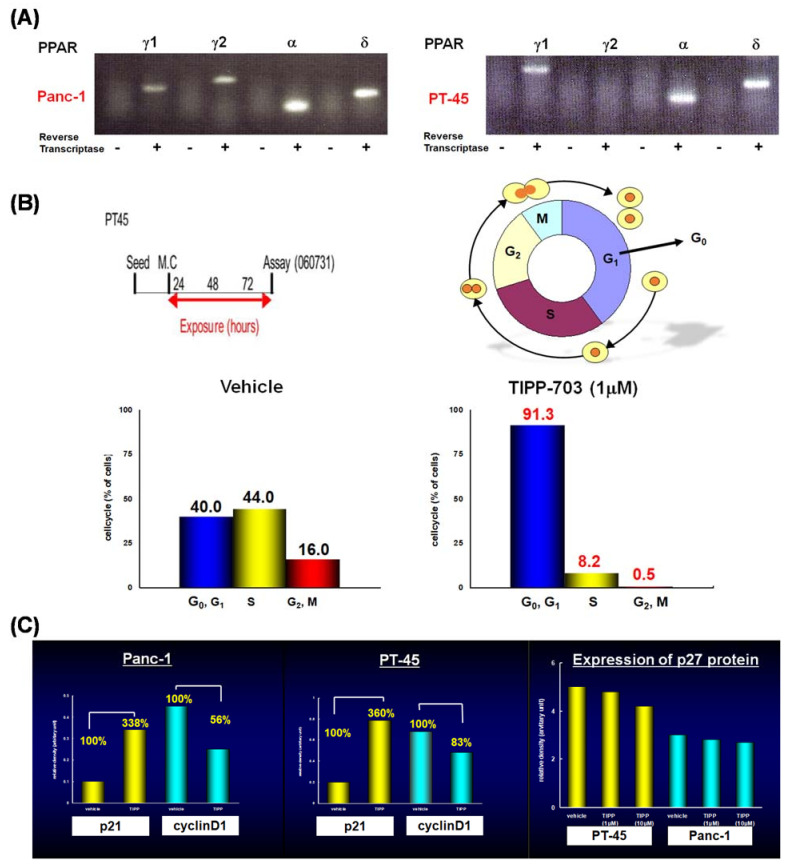
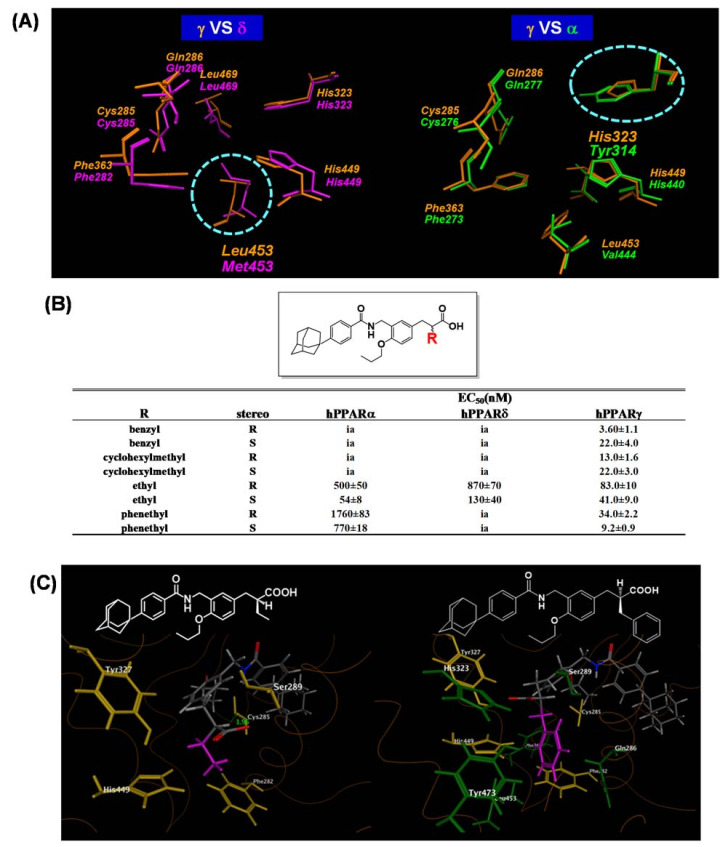
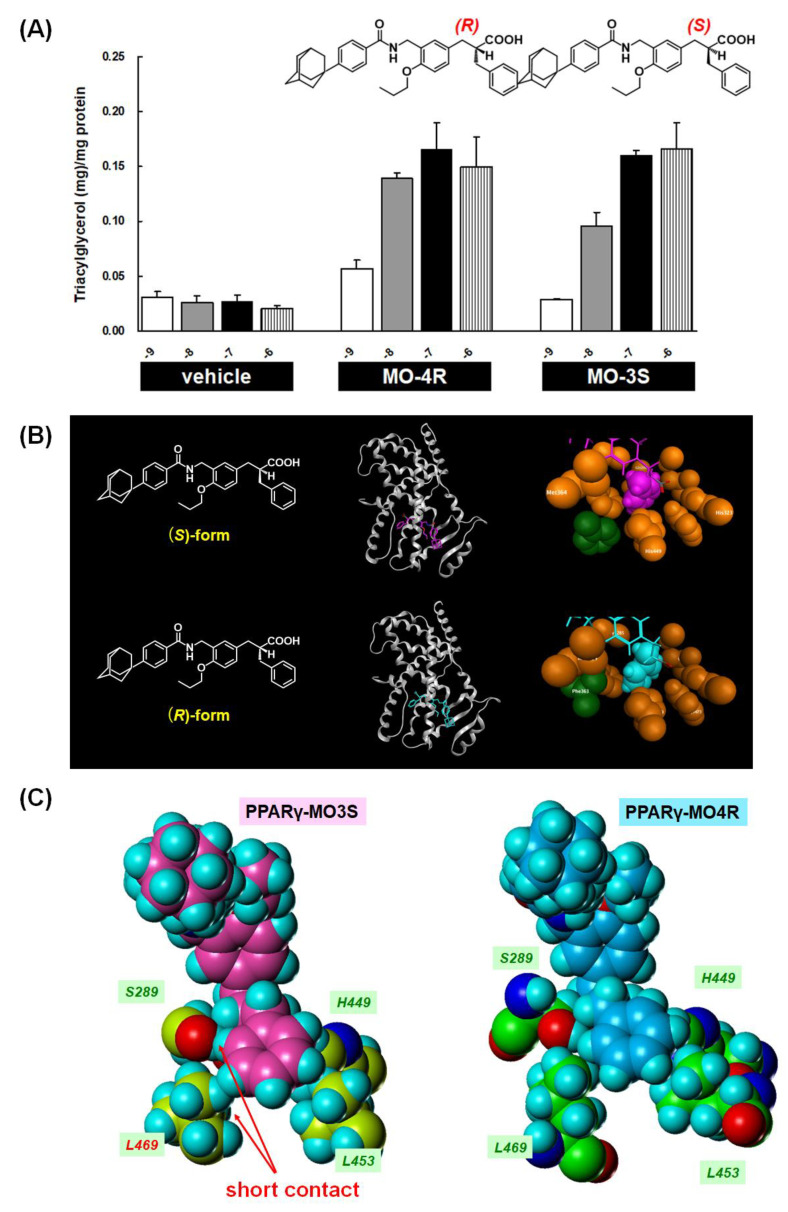
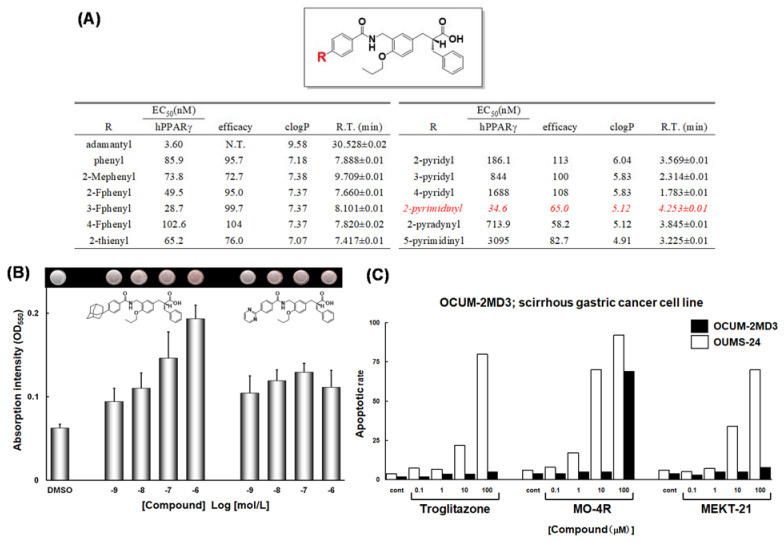
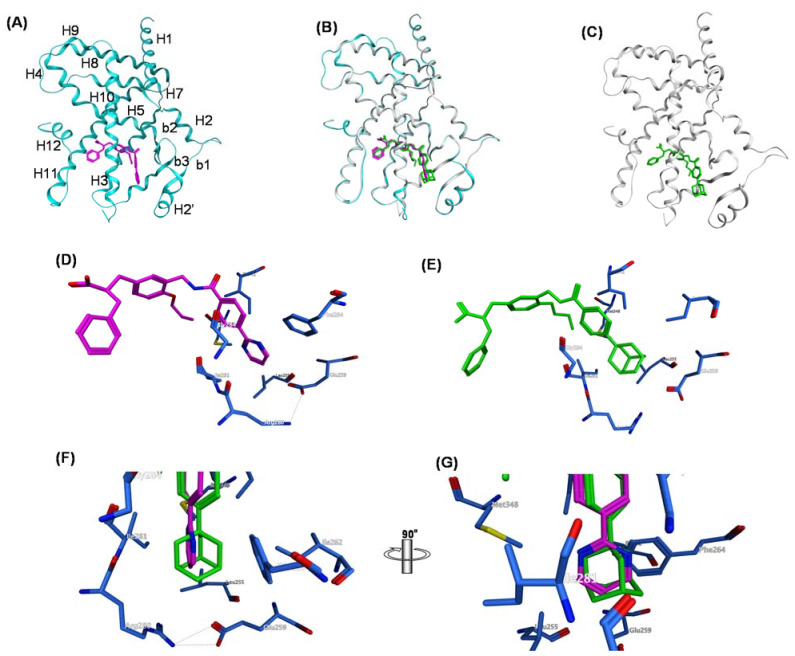
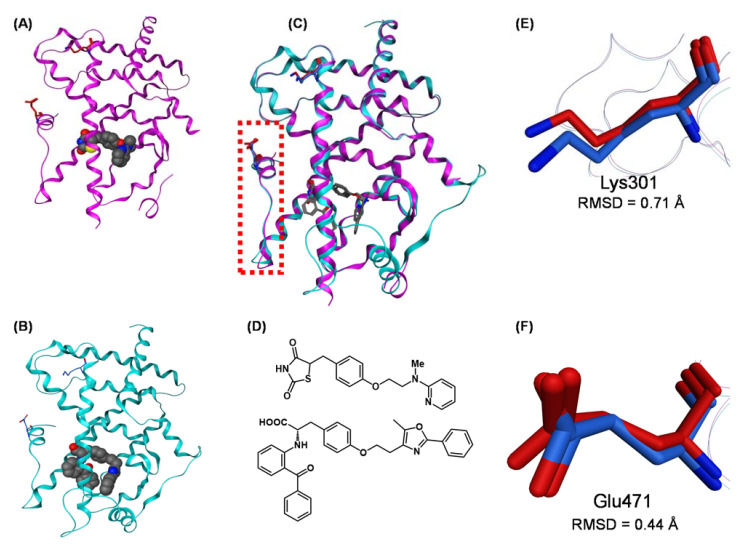

References
-
- Chawta A., Repa J.J., Evans R.M., Mangelsdorf D.J. Nuclear receptors and lipid physiology: Opening the x-files. Science. 2001;294:1866–1870. - PubMed
-
- Banner C.D., Gottlicher M., Widmark E., Sjovall J., Rafter J.J., Gustafsson J.A. A systematic analytical chemistry/cell assay approach to isolate activators of orphan nuclear receptors from biological extracts: Characterization of peroxisome proliferator-activated receptor activators in plasma. J. Lipid Res. 1993;34:1583–1591. doi: 10.1016/S0022-2275(20)36951-0. - DOI - PubMed
-
- Keller H., Dreyer C., Medin J., Mahfoudi A., Ozato K., Wahli W. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid X receptor heterodimers. Proc. Natl. Acad. Sci. USA. 1993;90:2160–2164. doi: 10.1073/pnas.90.6.2160. - DOI - PMC - PubMed
-
- Staels B., Auwerx J. Role of PPAR in the pharmacological regulation of lipoprotein metabolism by fibrates and thiazolidinediones. Curr. Pharm. Des. 1997;3:1–14.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
