

Kallikrein 5 Inhibition by the Lympho-Epithelial Kazal-Type Related Inhibitor Hinders Matriptase-Dependent Carcinogenesis
- PMID: 34503205
- PMCID: PMC8431081
- DOI: 10.3390/cancers13174395
Kallikrein 5 Inhibition by the Lympho-Epithelial Kazal-Type Related Inhibitor Hinders Matriptase-Dependent Carcinogenesis
Abstract
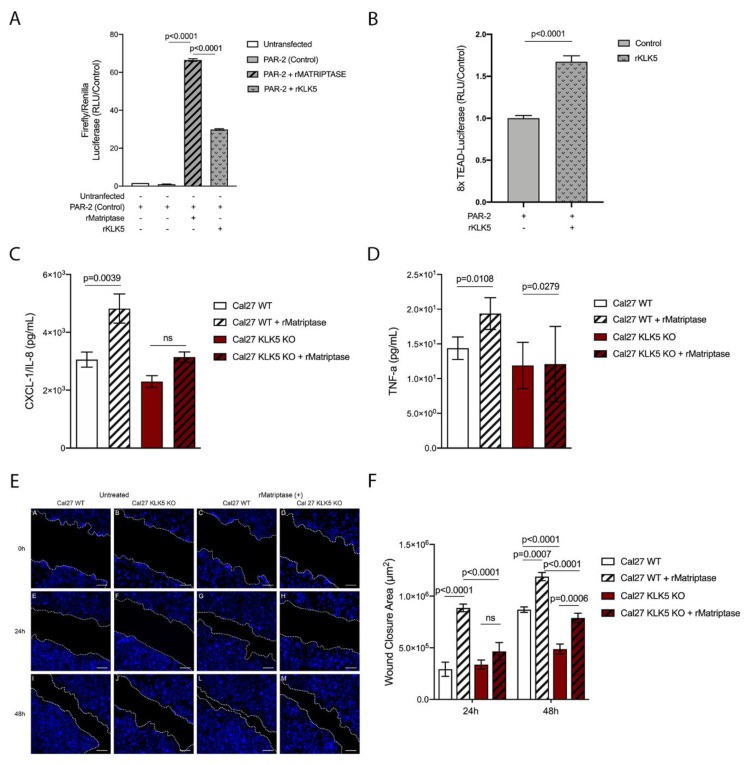
Head and neck squamous cell carcinoma remains challenging to treat with no improvement in survival rates over the past 50 years. Thus, there is an urgent need to discover more reliable therapeutic targets and biomarkers for HNSCC. Matriptase, a type-II transmembrane serine protease, induces malignant transformation in epithelial stem cells through proteolytic activation of pro-HGF and PAR-2, triggering PI3K-AKT-mTOR and NFKB signaling. The serine protease inhibitor lympho-epithelial Kazal-type-related inhibitor (LEKTI) inhibits the matriptase-driven proteolytic pathway, directly blocking kallikreins in epithelial differentiation. Hence, we hypothesized LEKTI could inhibit matriptase-dependent squamous cell carcinogenesis, thus implicating kallikreins in this process. Double-transgenic mice with simultaneous expression of matriptase and LEKTI under the keratin-5 promoter showed a prominent rescue of K5-Matriptase+/0 premalignant phenotype. Notably, in DMBA-induced SCC, heterotopic co-expression of LEKTI and matriptase delayed matriptase-driven tumor incidence and progression. Co-expression of LEKTI reverted altered Kallikrein-5 expression observed in the skin of K5-Matriptase+/0 mice, indicating that matriptase-dependent proteolytic pathway inhibition by LEKTI occurs through kallikreins. Moreover, we showed that Kallikrein-5 is necessary for PAR-2-mediated IL-8 release, YAP1-TAZ/TEAD activation, and matriptase-mediated oral squamous cell carcinoma migration. Collectively, our data identify a third signaling pathway for matriptase-dependent carcinogenesis in vivo. These findings are critical for the identification of more reliable biomarkers and effective therapeutic targets in Head and Neck cancer.
Keywords: KLK5; LEKTI; OSCC; SPINK5; matriptase.
Conflict of interest statement
The authors declare no potential conflict of interest.
Figures
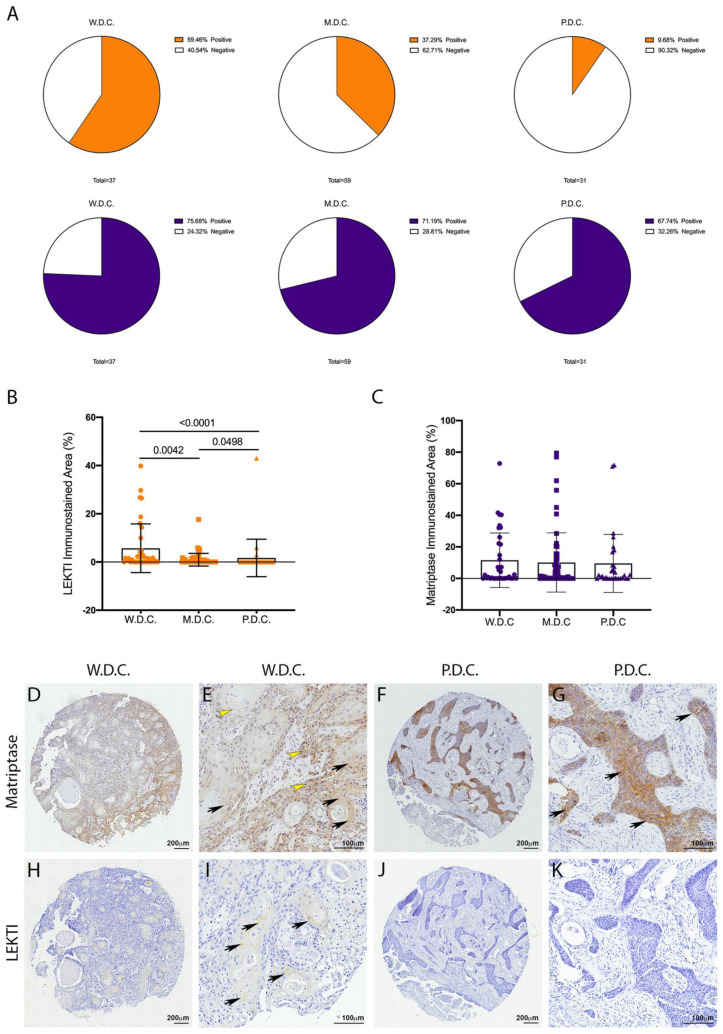
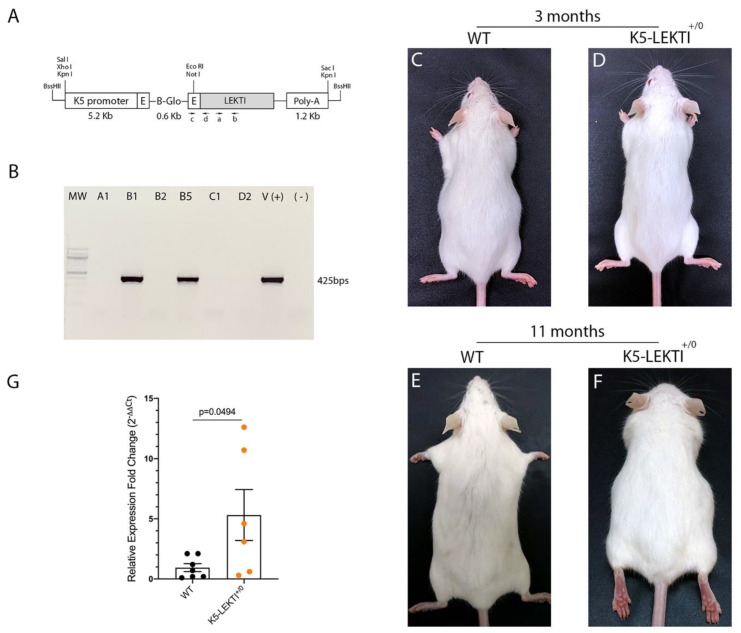
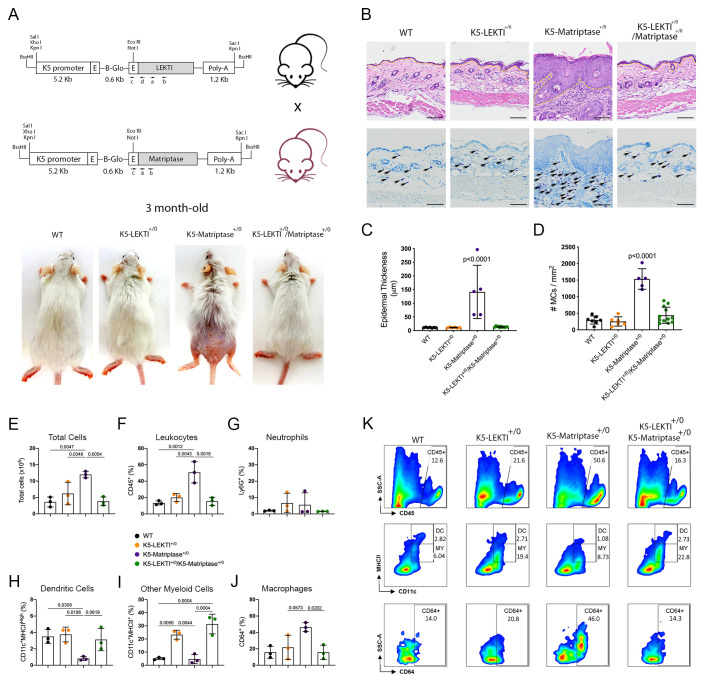
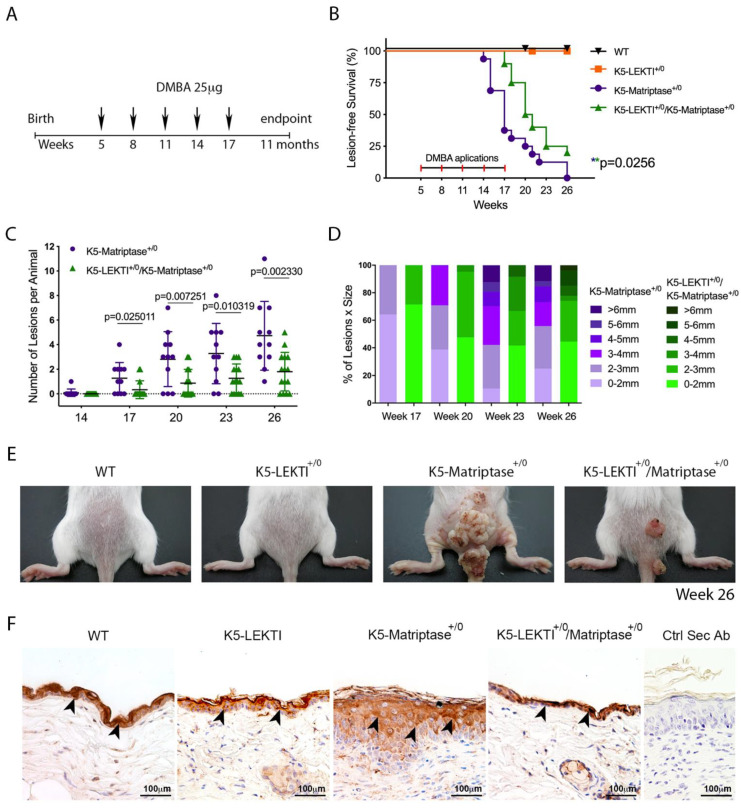
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
