

Molecular Mechanisms of Alcohol-Induced Colorectal Carcinogenesis
- PMID: 34503214
- PMCID: PMC8431530
- DOI: 10.3390/cancers13174404
Molecular Mechanisms of Alcohol-Induced Colorectal Carcinogenesis
Abstract
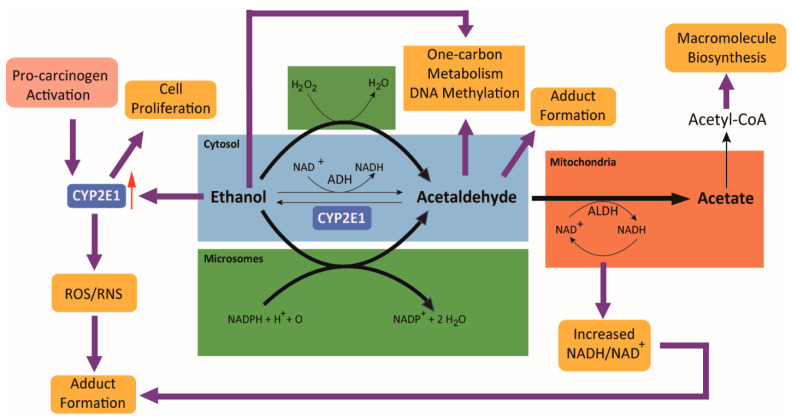
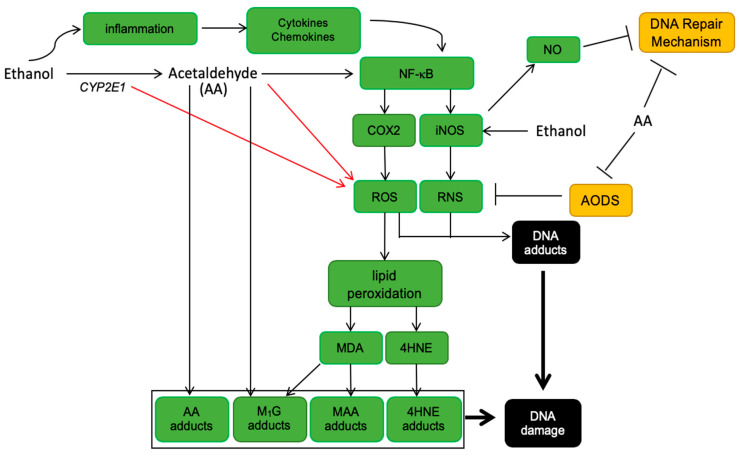
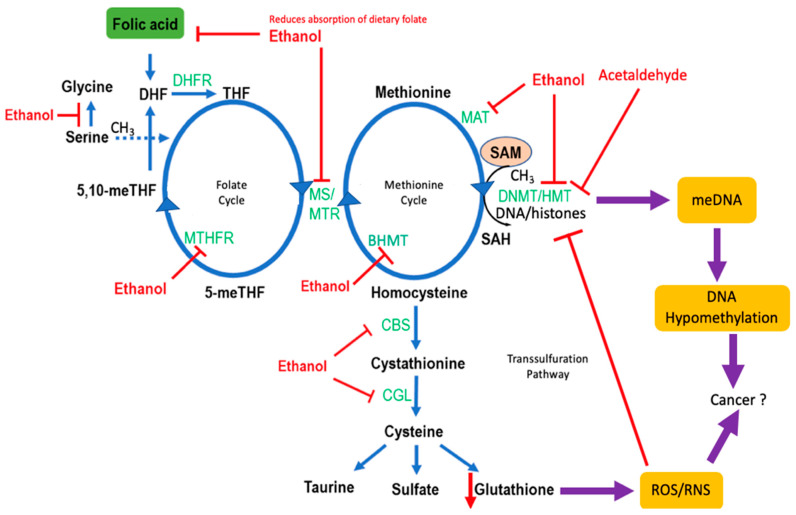
The etiology of colorectal cancer (CRC) is complex. Approximately, 10% of individuals with CRC have predisposing germline mutations that lead to familial cancer syndromes, whereas most CRC patients have sporadic cancer resulting from a combination of environmental and genetic risk factors. It has become increasingly clear that chronic alcohol consumption is associated with the development of sporadic CRC; however, the exact mechanisms by which alcohol contributes to colorectal carcinogenesis are largely unknown. Several proposed mechanisms from studies in CRC models suggest that alcohol metabolites and/or enzymes associated with alcohol metabolism alter cellular redox balance, cause DNA damage, and epigenetic dysregulation. In addition, alcohol metabolites can cause a dysbiotic colorectal microbiome and intestinal permeability, resulting in bacterial translocation, inflammation, and immunosuppression. All of these effects can increase the risk of developing CRC. This review aims to outline some of the most significant and recent findings on the mechanisms of alcohol in colorectal carcinogenesis. We examine the effect of alcohol on the generation of reactive oxygen species, the development of genotoxic stress, modulation of one-carbon metabolism, disruption of the microbiome, and immunosuppression.
Keywords: ALDH1B1; CRC; CYP2E1; DNA damage; acetaldehyde; alcohol; carcinogenesis; immunosuppression; microbiome; oxidative stress.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Dejea C.M., Wick E.C., Hechenbleikner E.M., White J.R., Mark Welch J.L., Rossetti B.J., Peterson S.N., Snesrud E.C., Borisy G.G., Lazarev M., et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA. 2014;111:18321–18326. doi: 10.1073/pnas.1406199111. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
