

Toward best practice in cancer mutation detection with whole-genome and whole-exome sequencing
- PMID: 34504346
- PMCID: PMC8506910
- DOI: 10.1038/s41587-021-00994-5
Toward best practice in cancer mutation detection with whole-genome and whole-exome sequencing
Abstract
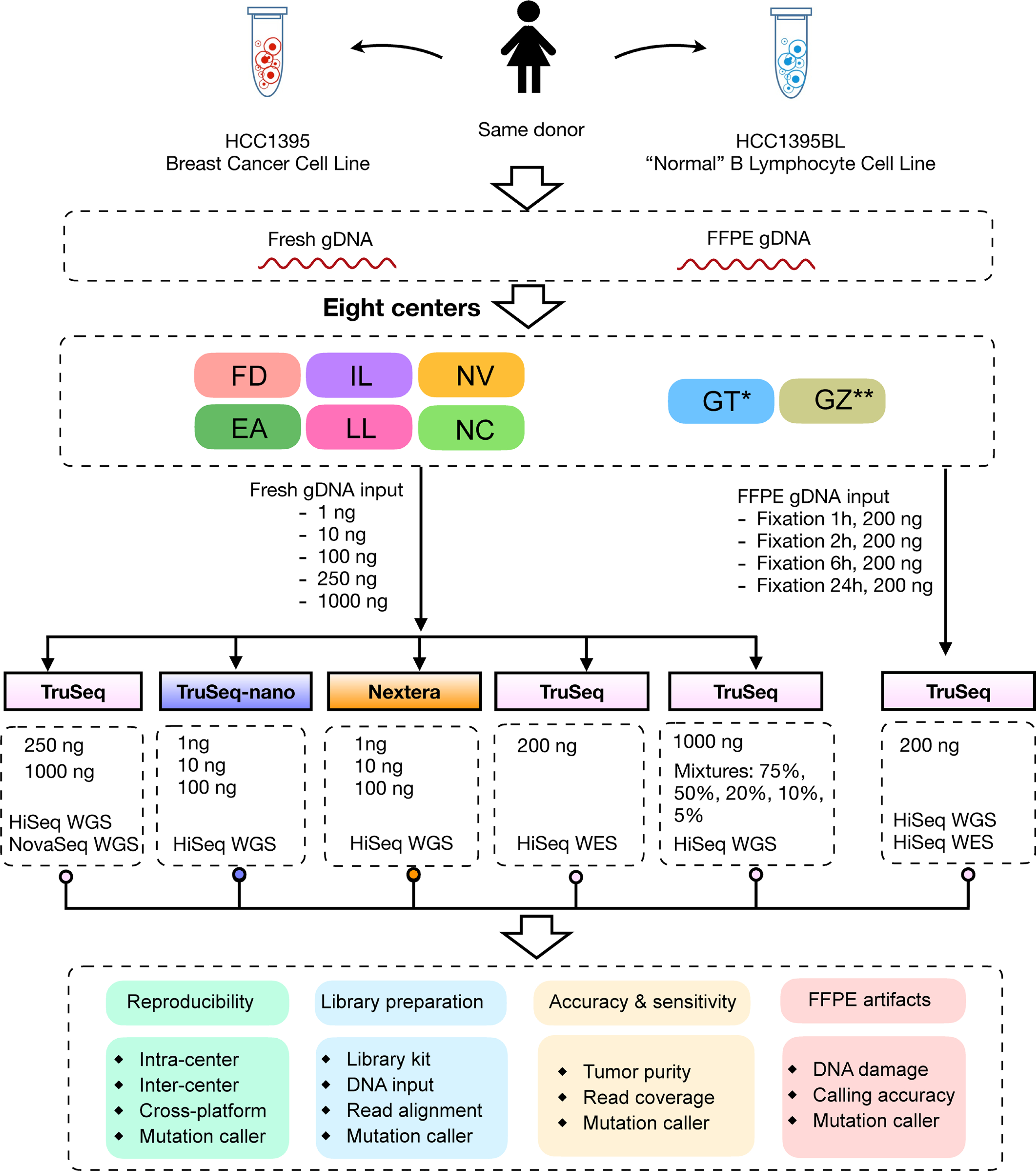
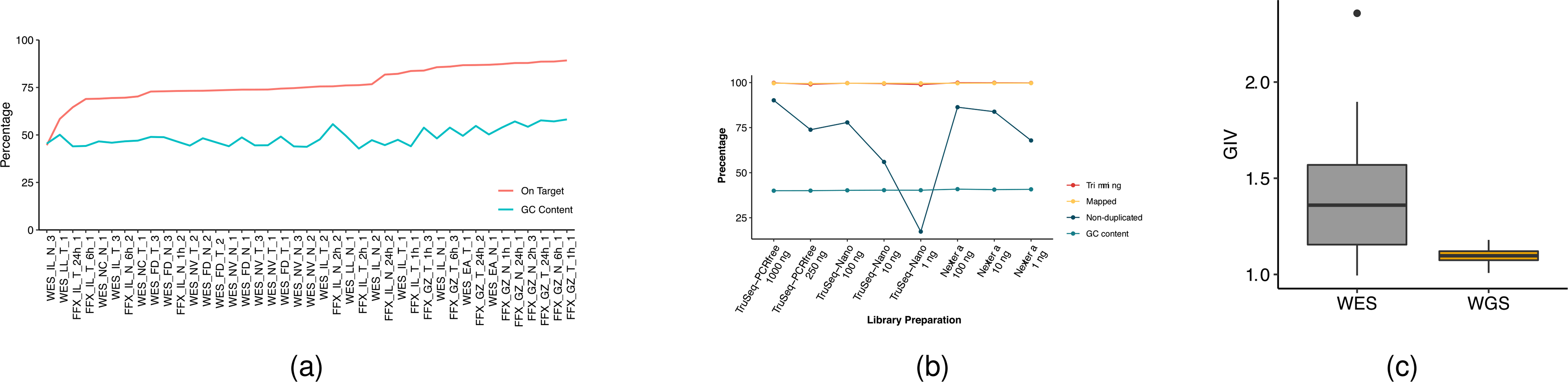
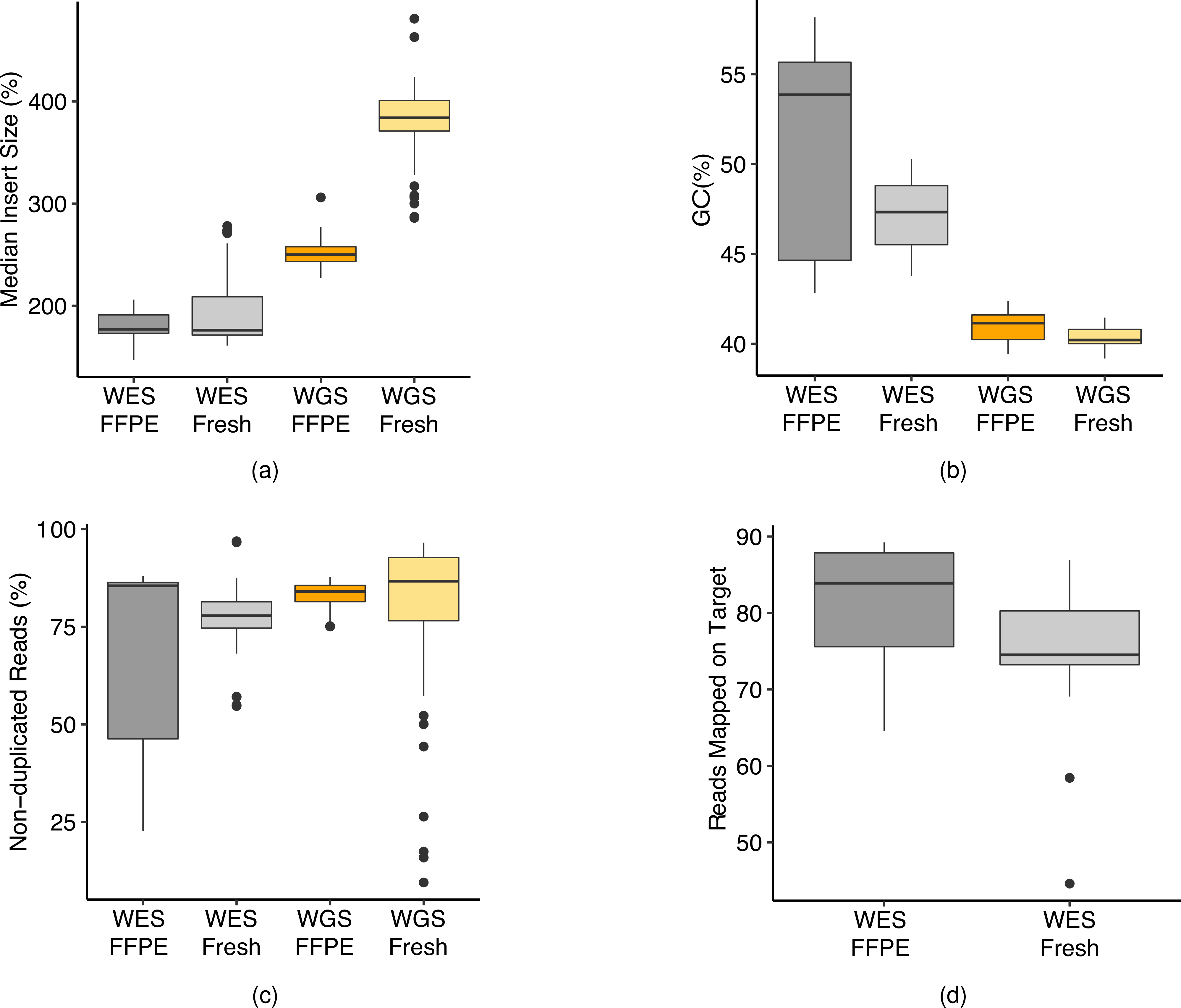
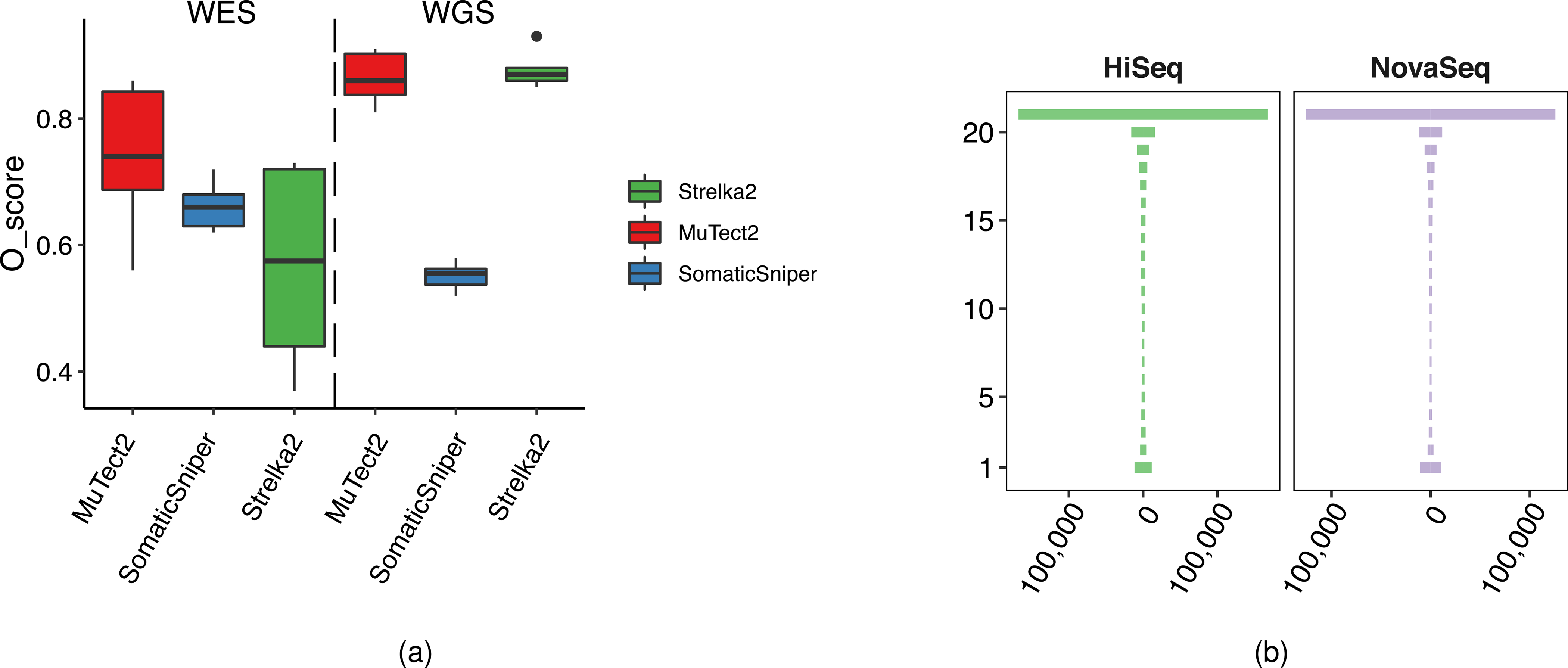
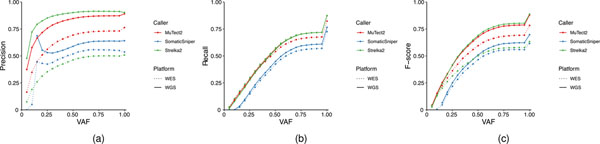
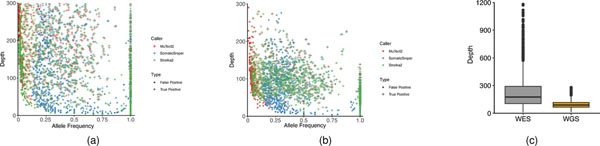
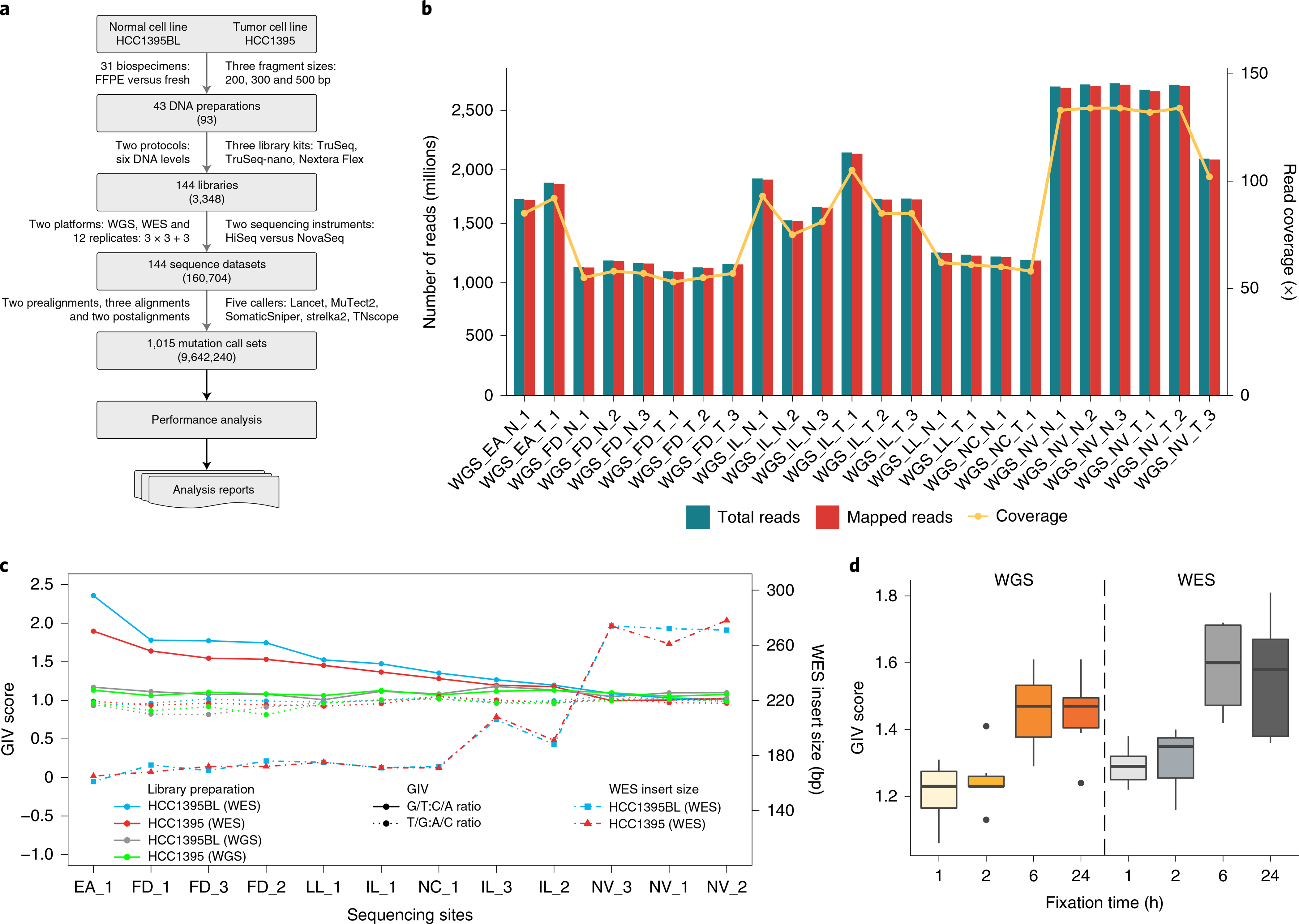
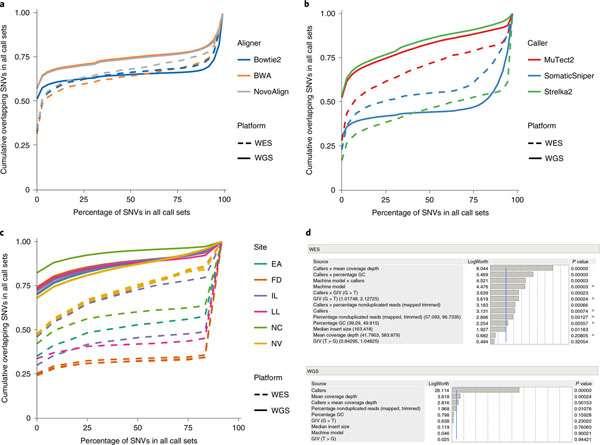
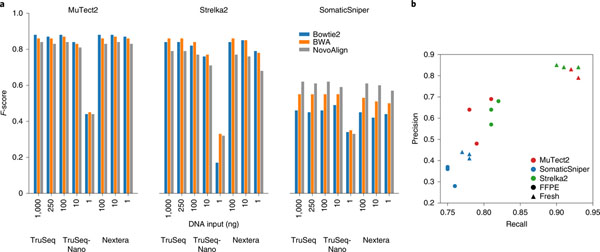
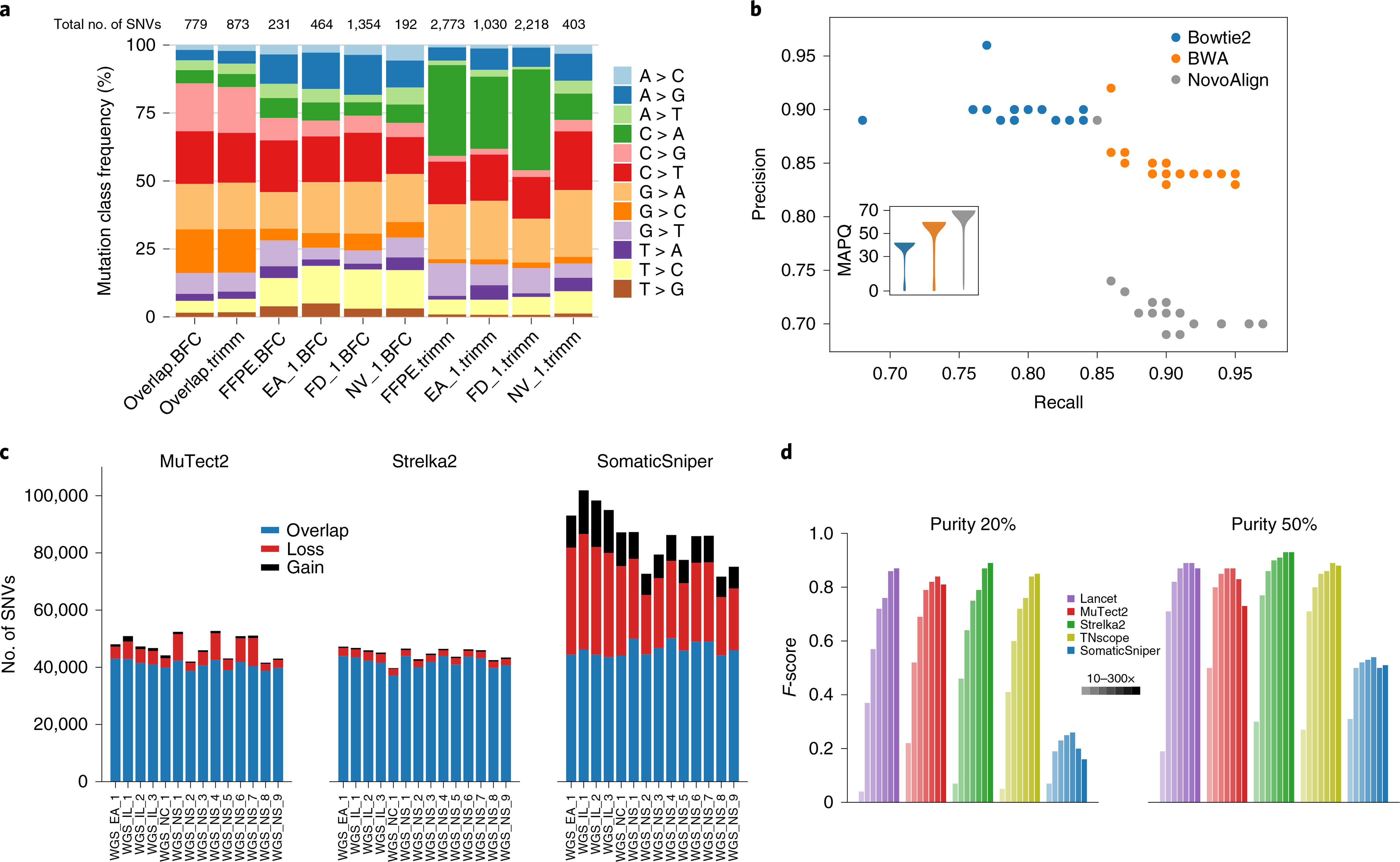
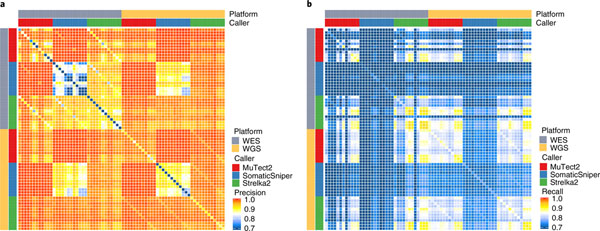
Clinical applications of precision oncology require accurate tests that can distinguish true cancer-specific mutations from errors introduced at each step of next-generation sequencing (NGS). To date, no bulk sequencing study has addressed the effects of cross-site reproducibility, nor the biological, technical and computational factors that influence variant identification. Here we report a systematic interrogation of somatic mutations in paired tumor-normal cell lines to identify factors affecting detection reproducibility and accuracy at six different centers. Using whole-genome sequencing (WGS) and whole-exome sequencing (WES), we evaluated the reproducibility of different sample types with varying input amount and tumor purity, and multiple library construction protocols, followed by processing with nine bioinformatics pipelines. We found that read coverage and callers affected both WGS and WES reproducibility, but WES performance was influenced by insert fragment size, genomic copy content and the global imbalance score (GIV; G > T/C > A). Finally, taking into account library preparation protocol, tumor content, read coverage and bioinformatics processes concomitantly, we recommend actionable practices to improve the reproducibility and accuracy of NGS experiments for cancer mutation detection.
© 2021. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
Competing interests
L.F. was an employees of Roche Sequencing Solutions Inc. L.K., K.L. and M.M. are employees of ATCC, which provided cell lines and derivative materials. E.J., O.D.A., T.T., A.M., A.N., A.G. and G.P.S. are employees of Illumina Inc. V.P. and M.S. are employees of Novartis Institutes for Biomedical Research. T.H., E.P and R. Kalamegham are employees of Genentech (a member of the Roche group). Z.L. is an employee of Sentieon Inc. R.K. is an employee of Immuneering Corp. C.E.M. is a cofounder of Onegevity Health. All other authors claim no competing interests.
Figures
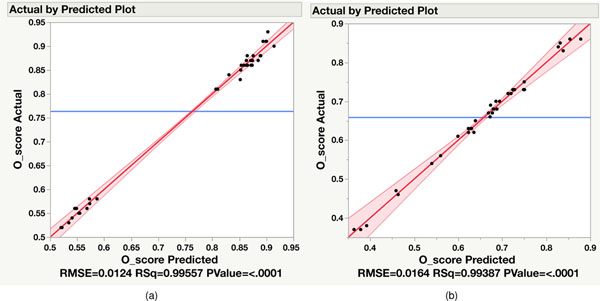
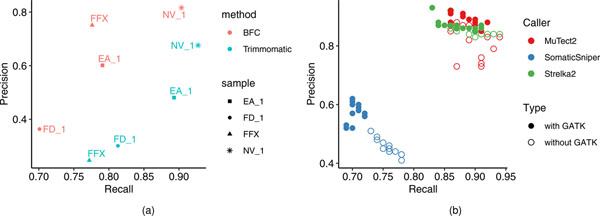
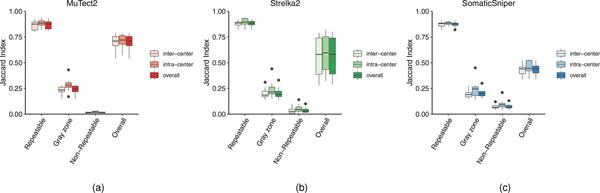
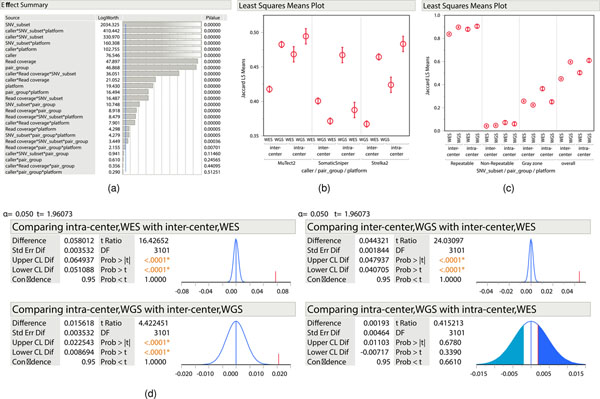
References
-
- Begley CG & Ellis LM Drug development: raise standards for preclinical cancer research. Nature 483, 531–533 (2012). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
