

Independent transcriptomic and proteomic regulation by type I and II protein arginine methyltransferases
- PMID: 34505004
- PMCID: PMC8417332
- DOI: 10.1016/j.isci.2021.102971
Independent transcriptomic and proteomic regulation by type I and II protein arginine methyltransferases
Abstract
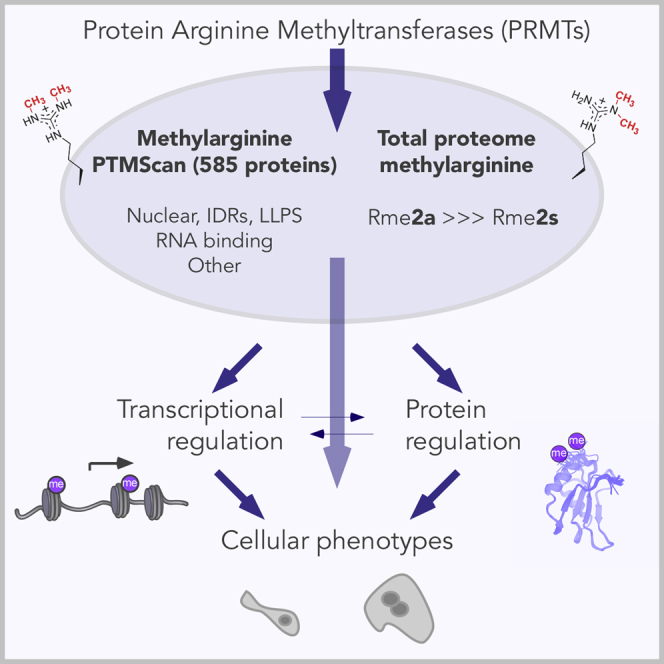
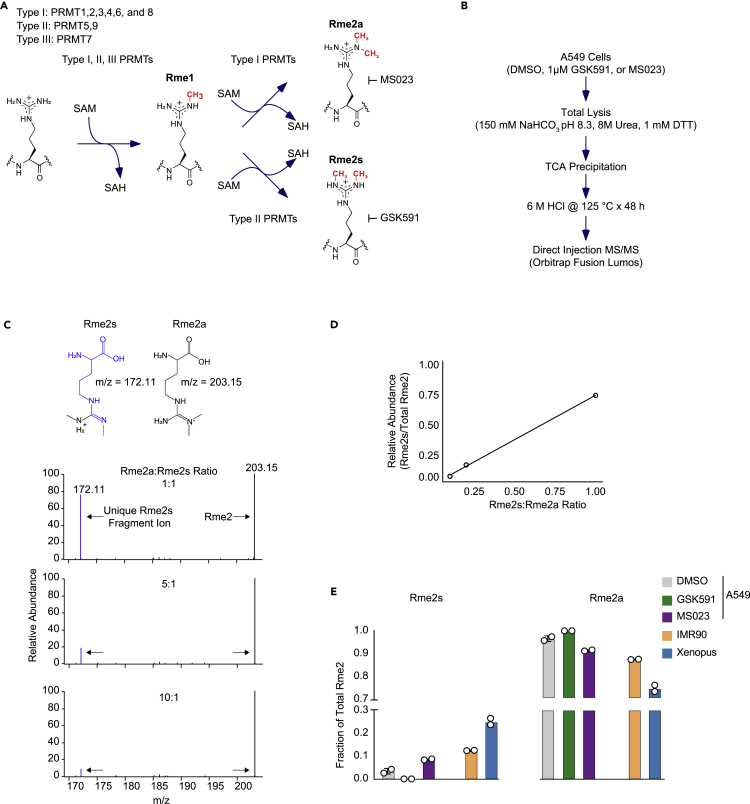
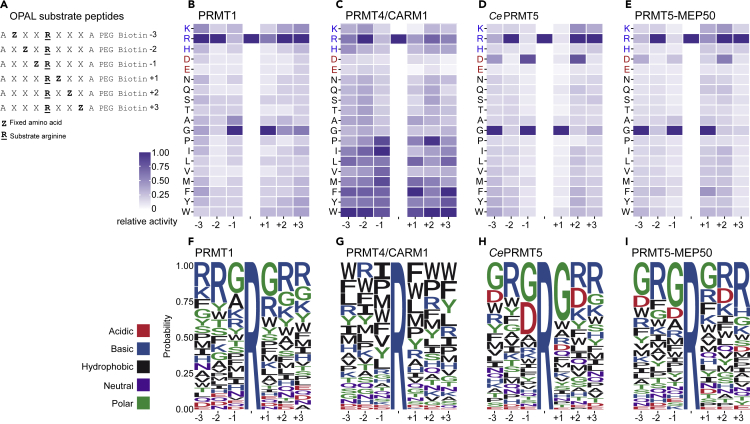
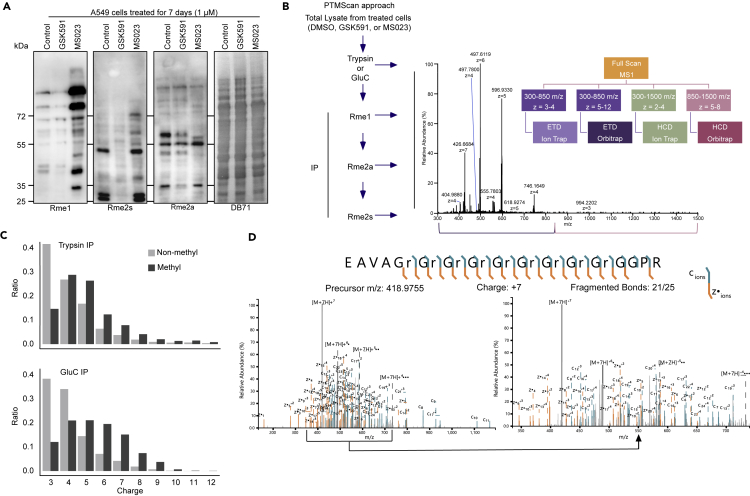
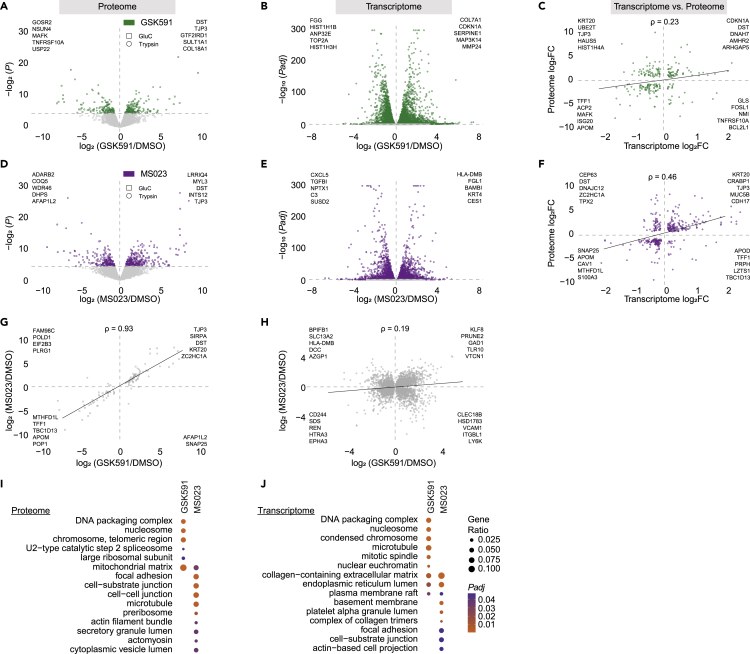
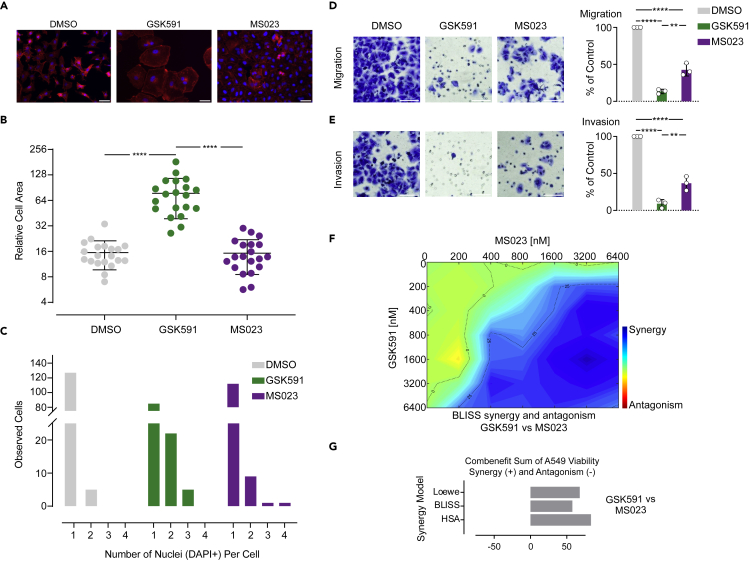
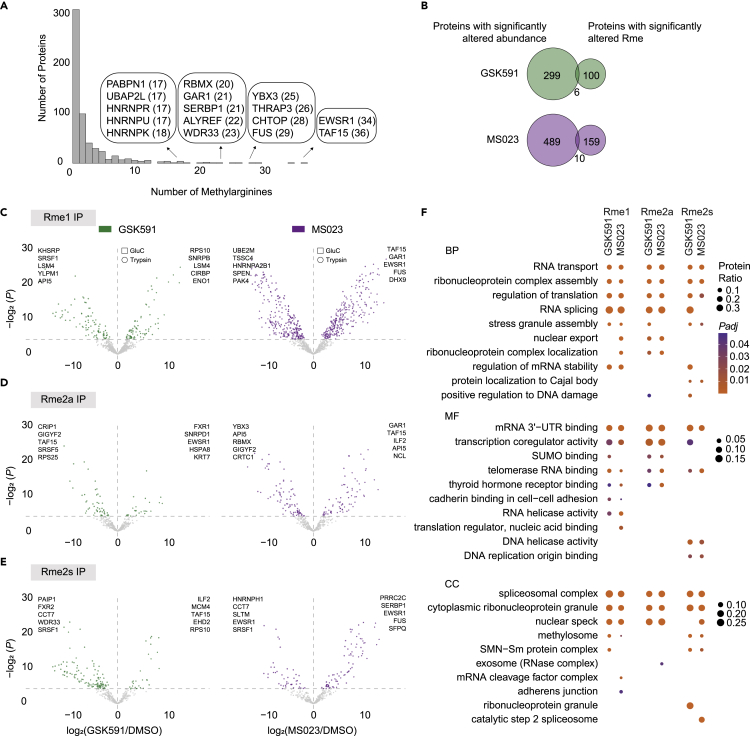
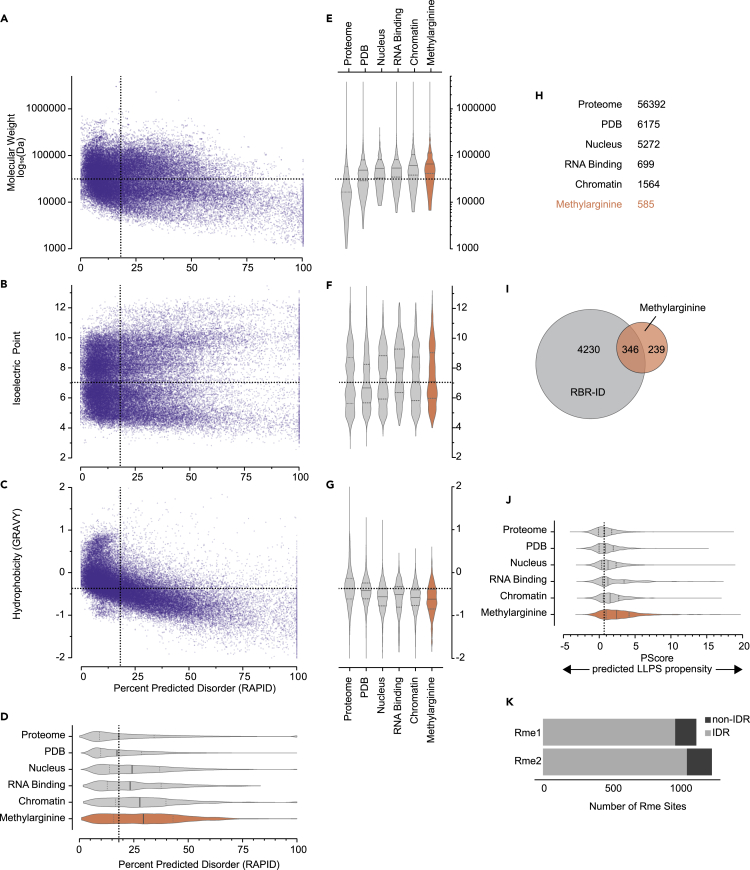
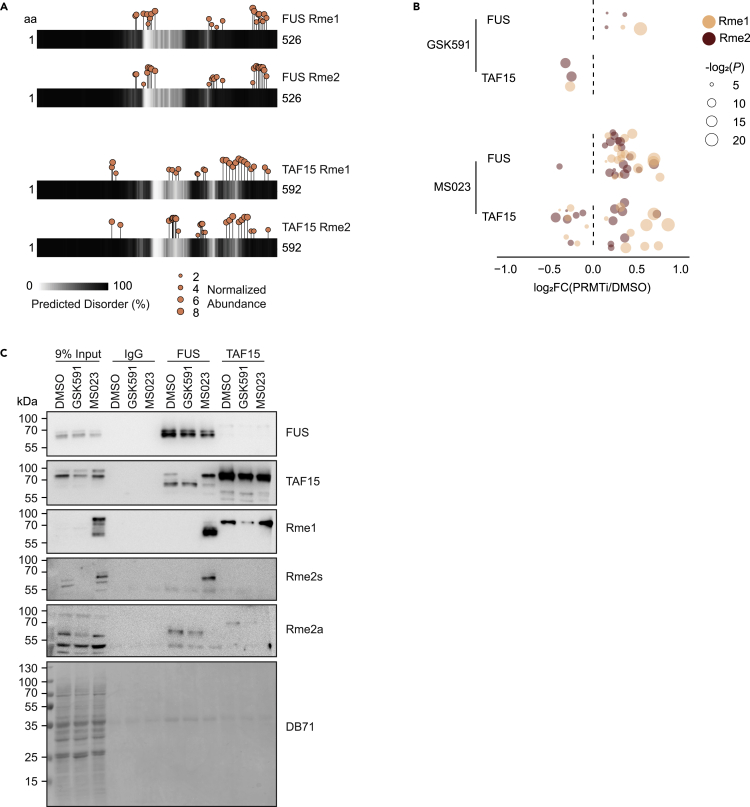
Protein arginine methyltransferases (PRMTs) catalyze the post-translational monomethylation (Rme1), asymmetric (Rme2a), or symmetric (Rme2s) dimethylation of arginine. To determine the cellular consequences of type I (Rme2a) and II (Rme2s) PRMTs, we developed and integrated multiple approaches. First, we determined total cellular dimethylarginine levels, revealing that Rme2s was ∼3% of total Rme2 and that this percentage was dependent upon cell type and PRMT inhibition status. Second, we quantitatively characterized in vitro substrates of the major enzymes and expanded upon PRMT substrate recognition motifs. We also compiled our data with publicly available methylarginine-modified residues into a comprehensive database. Third, we inhibited type I and II PRMTs and performed proteomic and transcriptomic analyses to reveal their phenotypic consequences. These experiments revealed both overlapping and independent PRMT substrates and cellular functions. Overall, this study expands upon PRMT substrate diversity, the arginine methylome, and the complex interplay of type I and II PRMTs.
Keywords: cell biology; molecular biology.
© 2021 The Author(s).
Conflict of interest statement
M.T.B. is a co-founder of EpiCypher.
Figures
References
-
- Barth T.K., Imhof A. Fast signals and slow marks: the dynamics of histone modifications. Trends Biochem. Sci. 2010;35:618–626. - PubMed
-
- Boffa L.C., Karn J., Vidali G., Allfrey V.G. Distribution of NG, NG,-dimethylarginine in nuclear protein fractions. Biochem. Biophys. Res. Commun. 1977;74:969–976. - PubMed
-
- Branscombe T.L., Frankel A., Lee J.H., Cook J.R., Yang Z., Pestka S., Clarke S. PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J. Biol. Chem. 2001;276:32971–32976. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
