

NetTCR-2.0 enables accurate prediction of TCR-peptide binding by using paired TCRα and β sequence data
- PMID: 34508155
- PMCID: PMC8433451
- DOI: 10.1038/s42003-021-02610-3
NetTCR-2.0 enables accurate prediction of TCR-peptide binding by using paired TCRα and β sequence data
Abstract
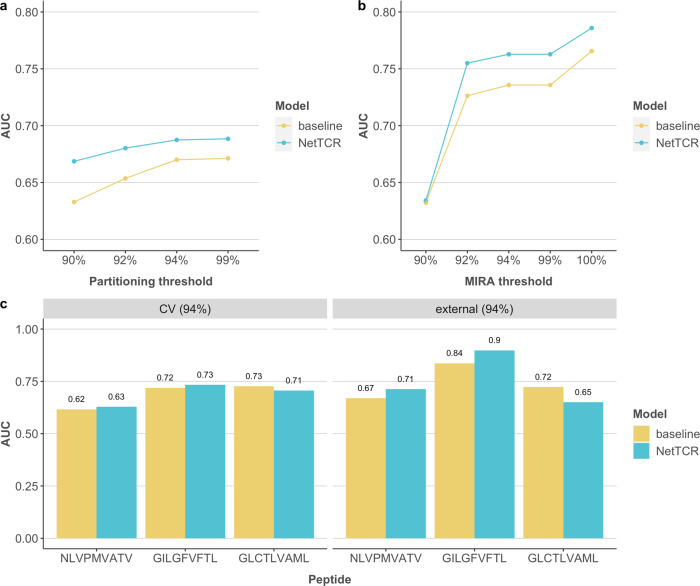
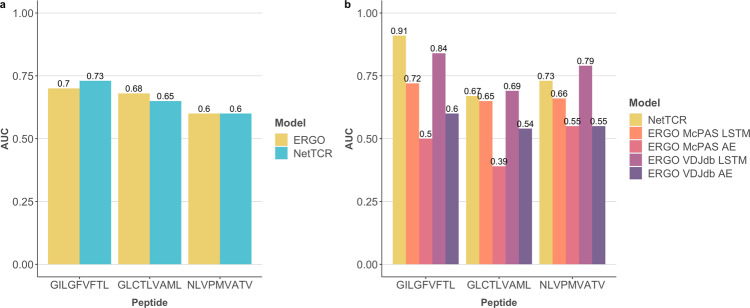
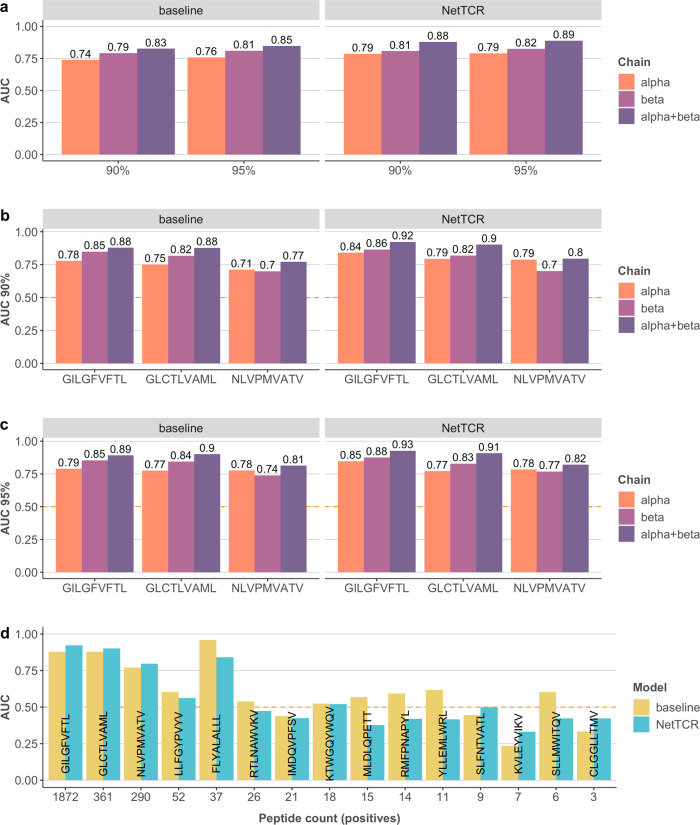
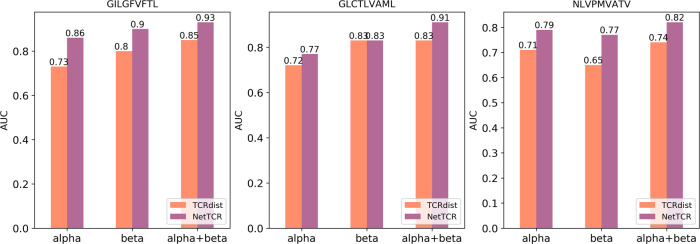
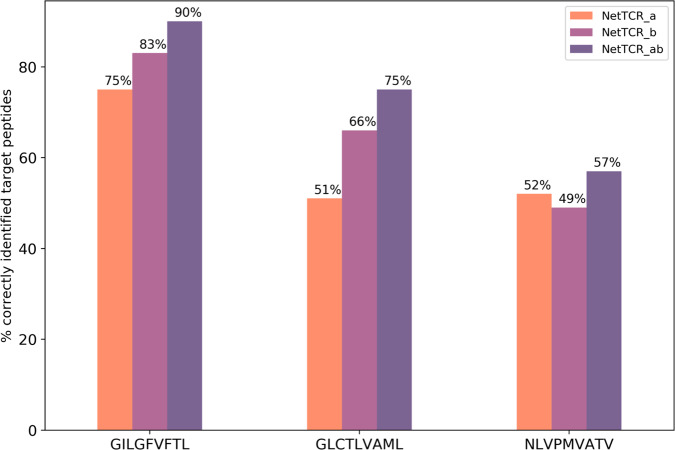
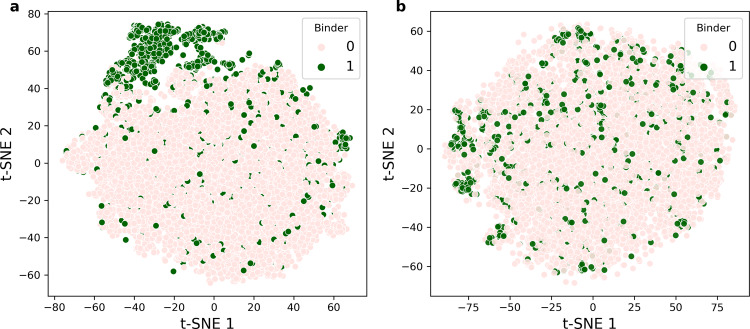
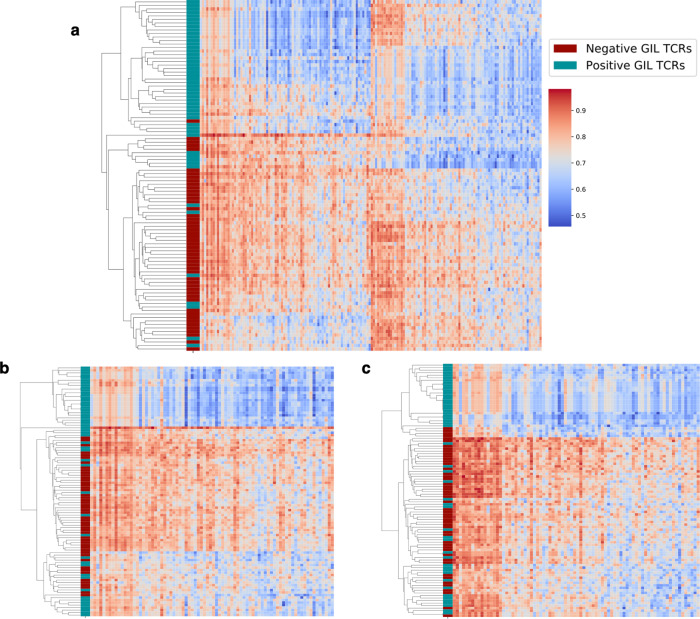
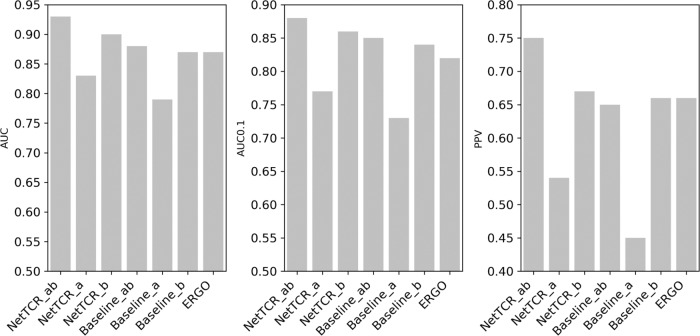
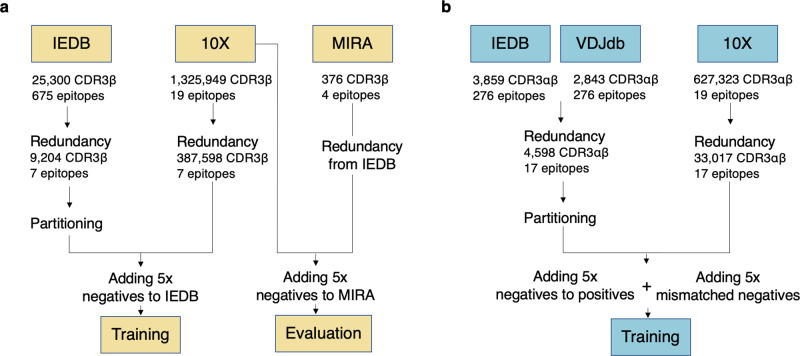
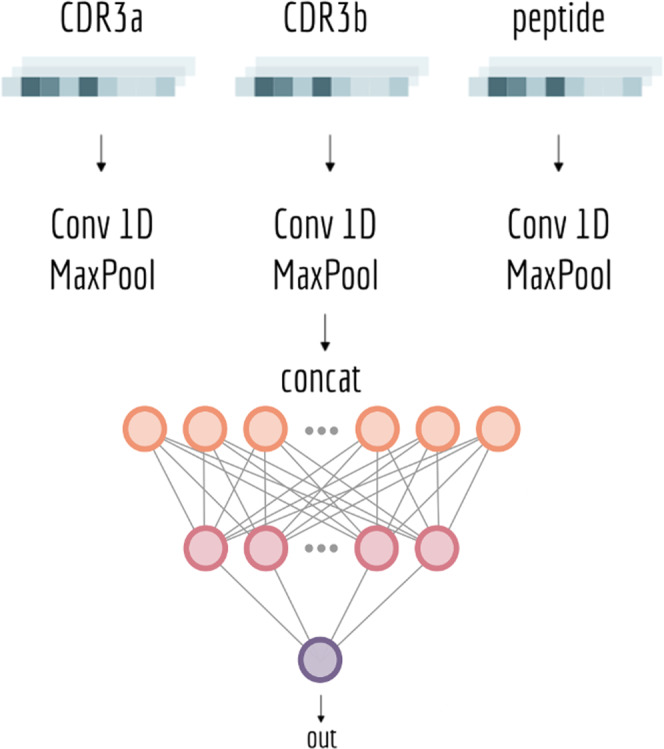
Prediction of T-cell receptor (TCR) interactions with MHC-peptide complexes remains highly challenging. This challenge is primarily due to three dominant factors: data accuracy, data scarceness, and problem complexity. Here, we showcase that "shallow" convolutional neural network (CNN) architectures are adequate to deal with the problem complexity imposed by the length variations of TCRs. We demonstrate that current public bulk CDR3β-pMHC binding data overall is of low quality and that the development of accurate prediction models is contingent on paired α/β TCR sequence data corresponding to at least 150 distinct pairs for each investigated pMHC. In comparison, models trained on CDR3α or CDR3β data alone demonstrated a variable and pMHC specific relative performance drop. Together these findings support that T-cell specificity is predictable given the availability of accurate and sufficient paired TCR sequence data. NetTCR-2.0 is publicly available at https://services.healthtech.dtu.dk/service.php?NetTCR-2.0 .
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
