

The MYC oncogene - the grand orchestrator of cancer growth and immune evasion
- PMID: 34508258
- PMCID: PMC9083341
- DOI: 10.1038/s41571-021-00549-2
The MYC oncogene - the grand orchestrator of cancer growth and immune evasion
Abstract
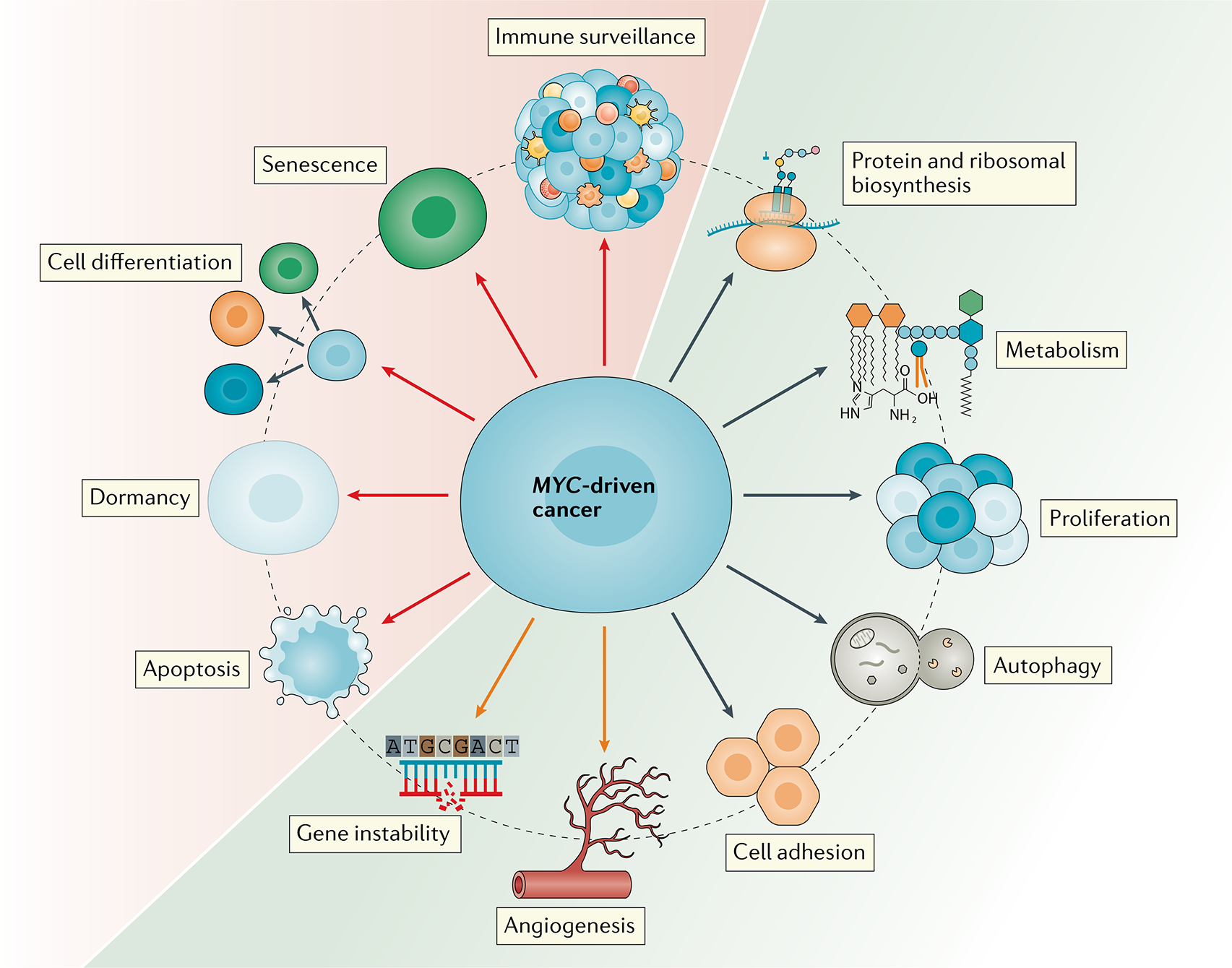
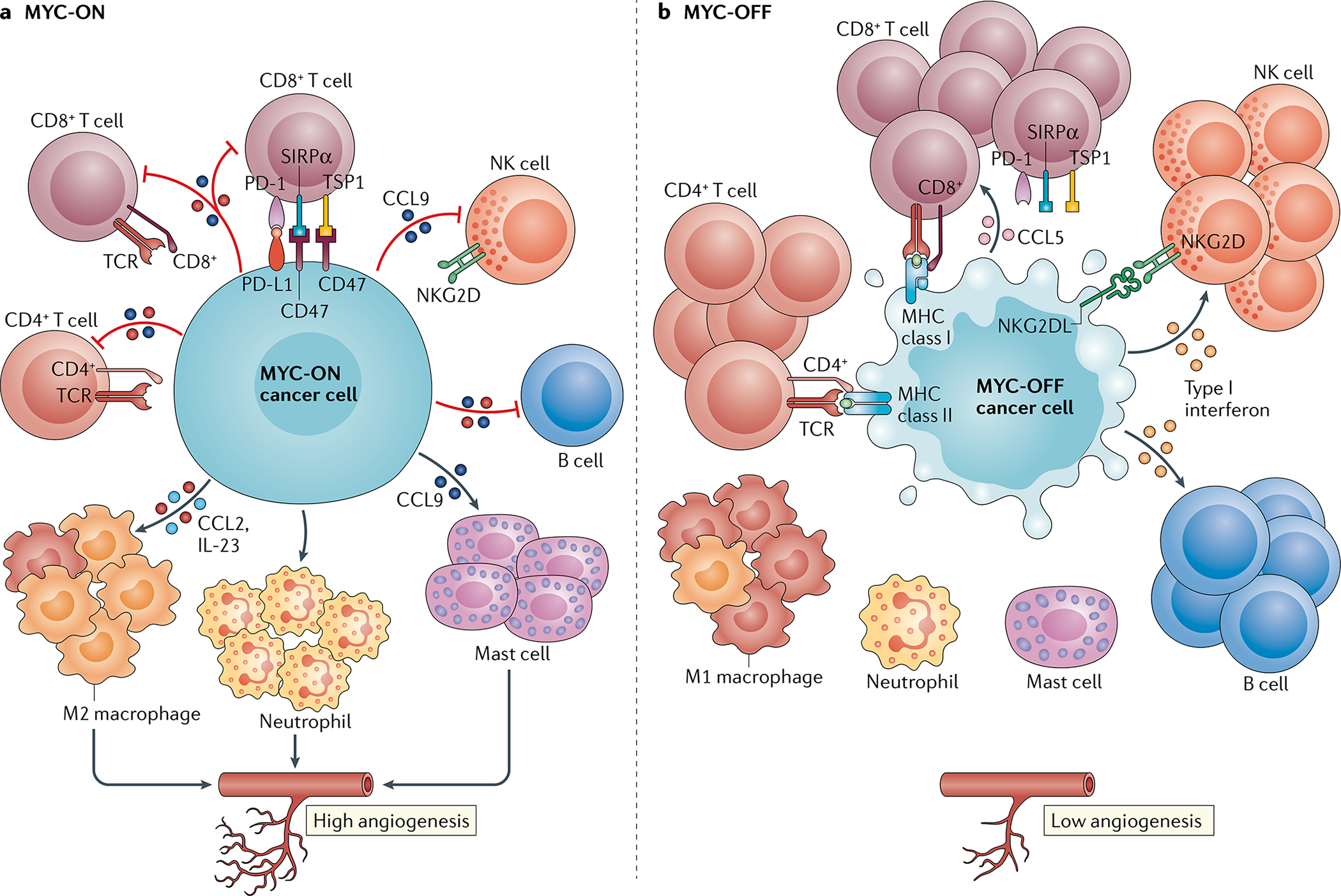
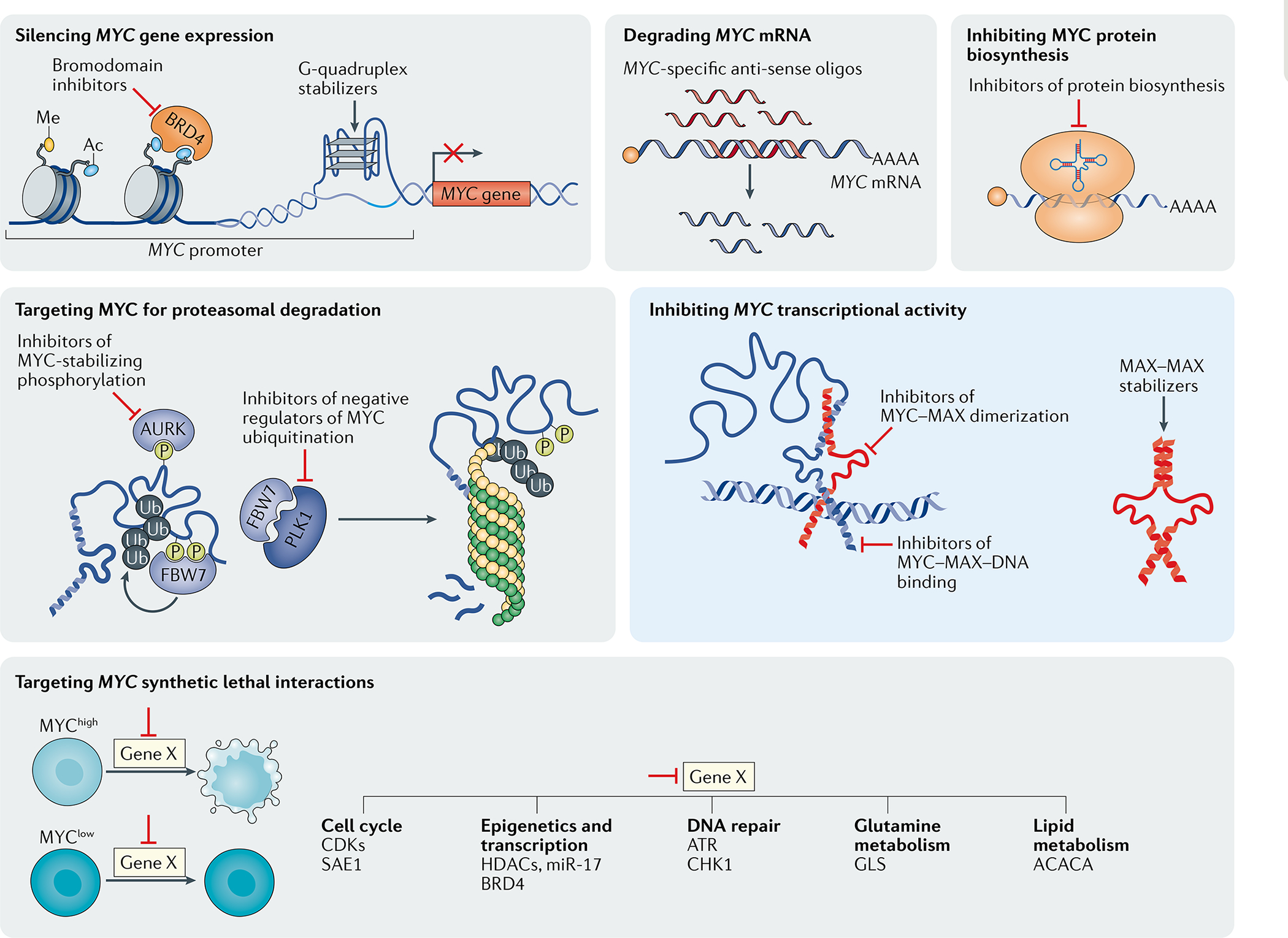
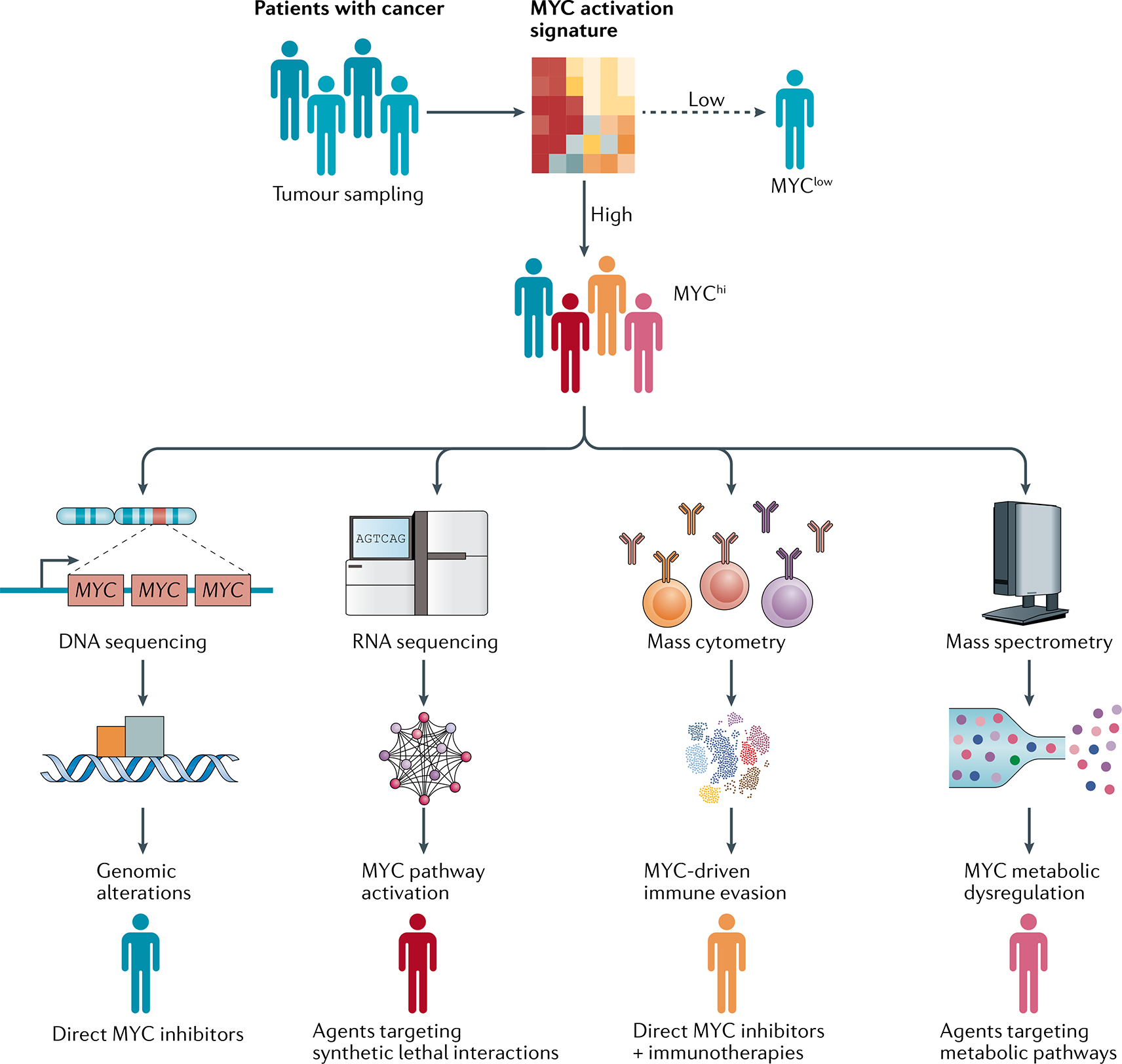
The MYC proto-oncogenes encode a family of transcription factors that are among the most commonly activated oncoproteins in human neoplasias. Indeed, MYC aberrations or upregulation of MYC-related pathways by alternate mechanisms occur in the vast majority of cancers. MYC proteins are master regulators of cellular programmes. Thus, cancers with MYC activation elicit many of the hallmarks of cancer required for autonomous neoplastic growth. In preclinical models, MYC inactivation can result in sustained tumour regression, a phenomenon that has been attributed to oncogene addiction. Many therapeutic agents that directly target MYC are under development; however, to date, their clinical efficacy remains to be demonstrated. In the past few years, studies have demonstrated that MYC signalling can enable tumour cells to dysregulate their microenvironment and evade the host immune response. Herein, we discuss how MYC pathways not only dictate cancer cell pathophysiology but also suppress the host immune response against that cancer. We also propose that therapies targeting the MYC pathway will be key to reversing cancerous growth and restoring antitumour immune responses in patients with MYC-driven cancers.
© 2021. Springer Nature Limited.
Conflict of interest statement
Competing interests
D.W.F. is a consultant for Revolution Medicines, a company developing MYC pathway therapies, co-founder of Bachus and Molecular Decisions, and has had advisory roles for American Gene Technologies, Geron, Moderna and Regulus. The other authors declare no conflicts of interest.
Figures
References
-
- Meyer N & Penn LZ Reflecting on 25 years with MYC. Nat. Rev. Cancer 8, 976–990 (2008). - PubMed
-
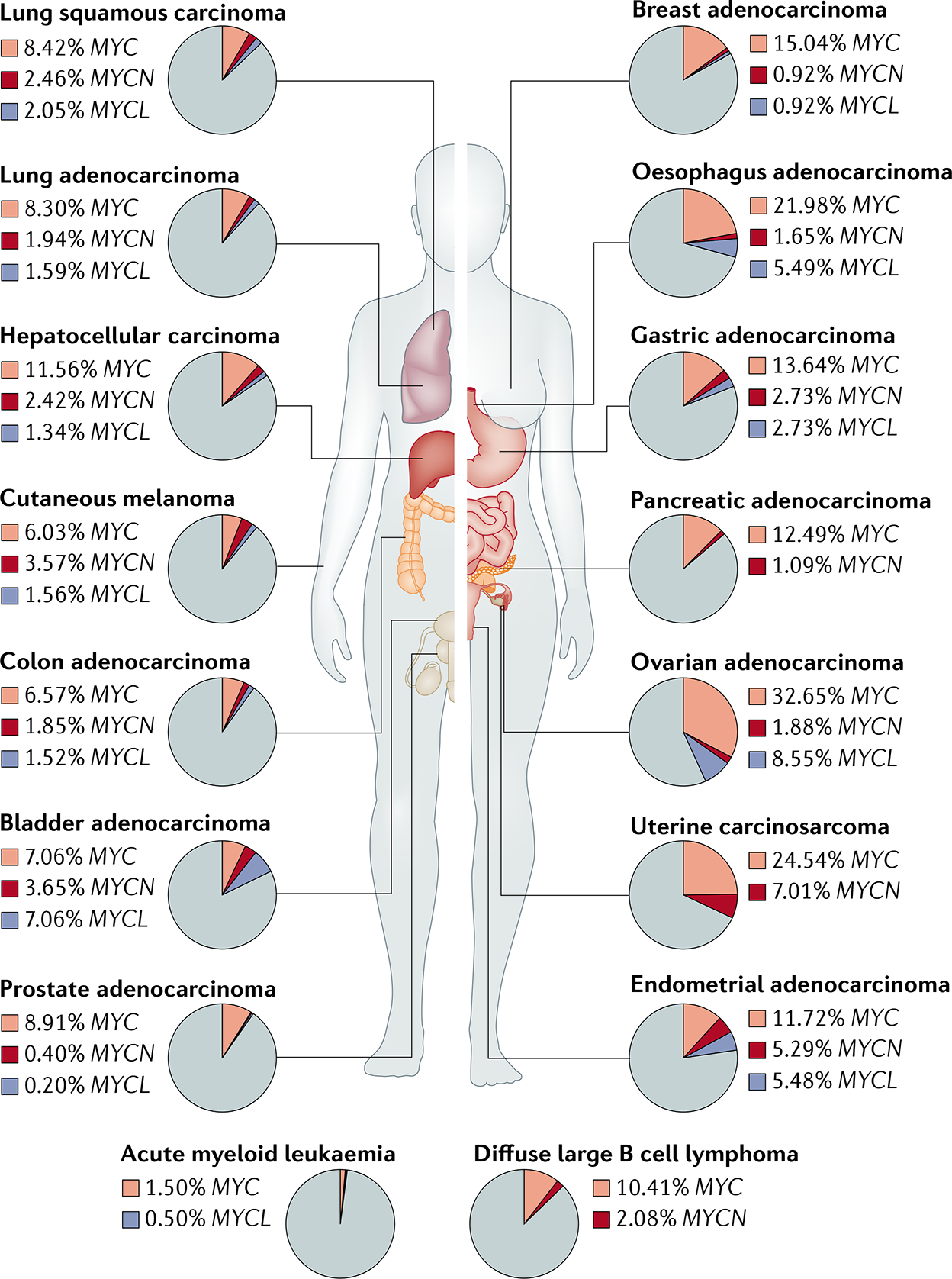
- Kalkat M et al. MYC deregulation in primary human cancers. Genes 8, 151 (2017).
-
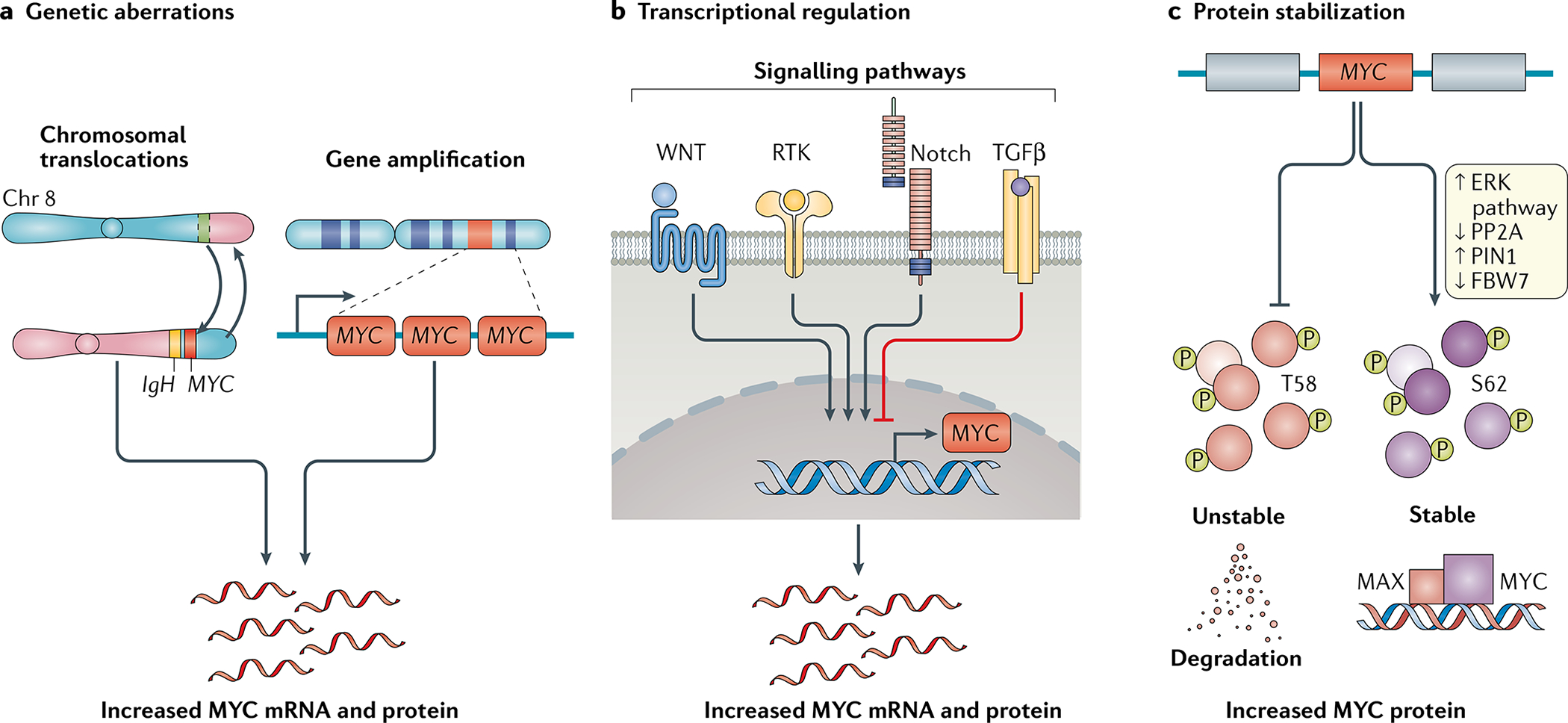
- Kress TR, Sabò A & Amati B MYC: connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 15, 593–607 (2015). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
