

Gene Therapy for Duchenne Muscular Dystrophy
- PMID: 34511510
- PMCID: PMC8673537
- DOI: 10.3233/JND-210678
Gene Therapy for Duchenne Muscular Dystrophy
Abstract
Duchenne muscular dystrophy (DMD) is an X-linked, muscle wasting disease that affects 1 in 5000 males. Affected individuals become wheelchair bound by the age of twelve and eventually die in their third decade due to respiratory and cardiac complications. The disease is caused by mutations in the DMD gene that codes for dystrophin. Dystrophin is a structural protein that maintains the integrity of muscle fibres and protects them from contraction-induced damage. The absence of dystrophin compromises the stability and function of the muscle fibres, eventually leading to muscle degeneration. So far, there is no effective treatment for deteriorating muscle function in DMD patients. A promising approach for treating this life-threatening disease is gene transfer to restore dystrophin expression using a safe, non-pathogenic viral vector called adeno-associated viral (AAV) vector. Whilst microdystrophin gene transfer using AAV vectors shows extremely impressive therapeutic success so far in large animal models of DMD, translating this advanced therapy medicinal product from bench to bedside still offers scope for many optimization steps. In this paper, the authors review the current progress of AAV-microdystrophin gene therapy for DMD and other treatment strategies that may apply to a subset of DMD patients depending on the mutations they carry.
Keywords: Gene therapy; adeno-associated virus; antisense; duchenne; dystrophin; exon skipping; microdystrophin; muscular dystrophy.
Conflict of interest statement
The authors have no conflict to report. GD has acted as a consultant for SynPromics (AskBio), Sarepta and RegeneX. GD is a named inventor on patents related to DMD gene therapy and exon skipping.
Figures
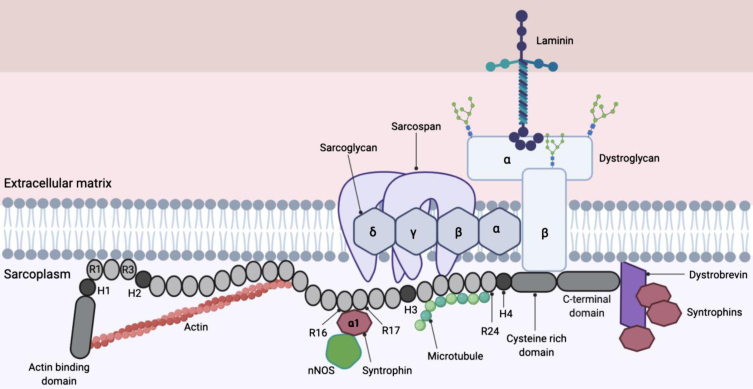
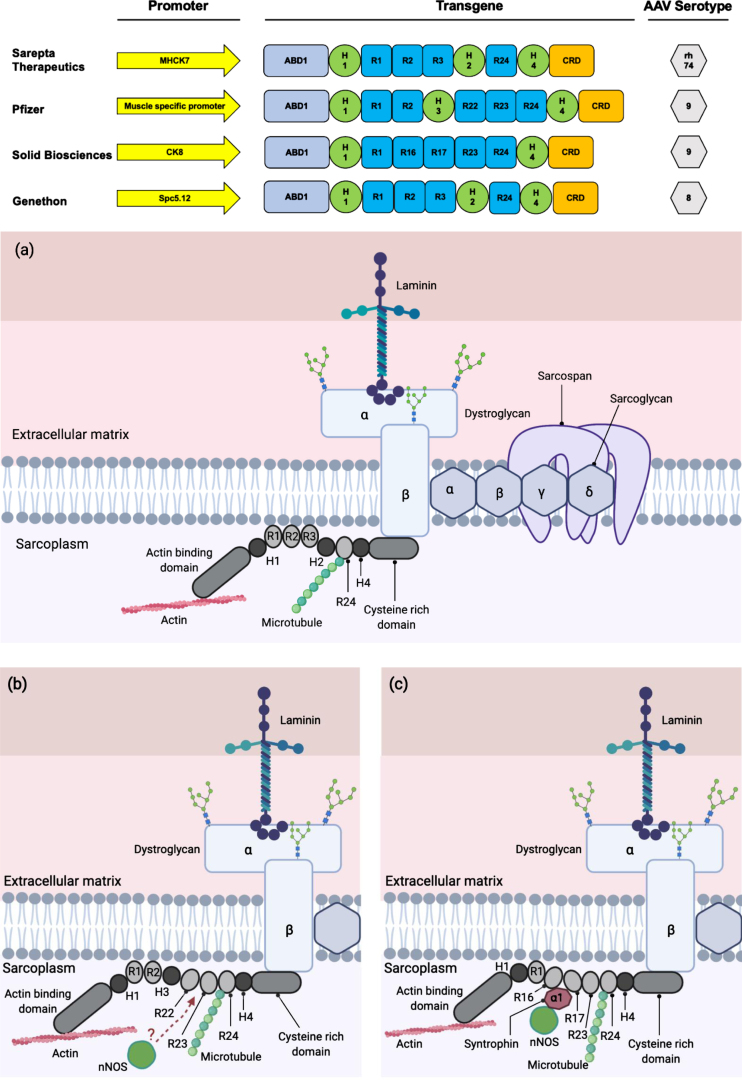
References
-
- Angelini C. Muscular dystrophy. Handbook of clinical neurology: Elsevier; 2009. p. 477–488. - PubMed
-
- Emery AE. The muscular dystrophies. The Lancet. 2002;359(9307):687–95. - PubMed
-
- Gowers WR. Pseudo-hypertrophic muscular paralysis: a clinical lecture. : J. & A. Churchill; 1879.
-
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509–17. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
