

A statistical model for describing and simulating microbial community profiles
- PMID: 34516542
- PMCID: PMC8491899
- DOI: 10.1371/journal.pcbi.1008913
A statistical model for describing and simulating microbial community profiles
Abstract
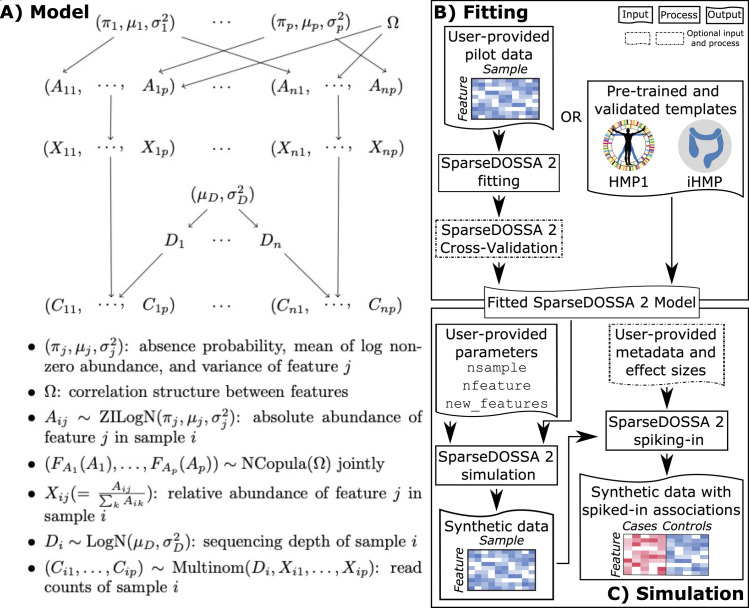
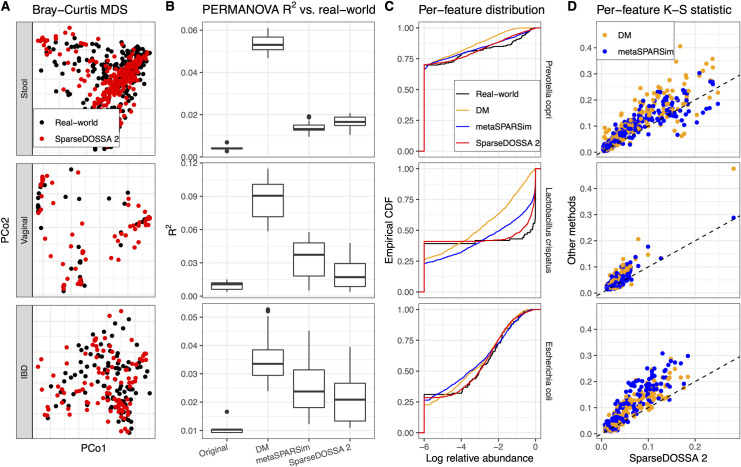
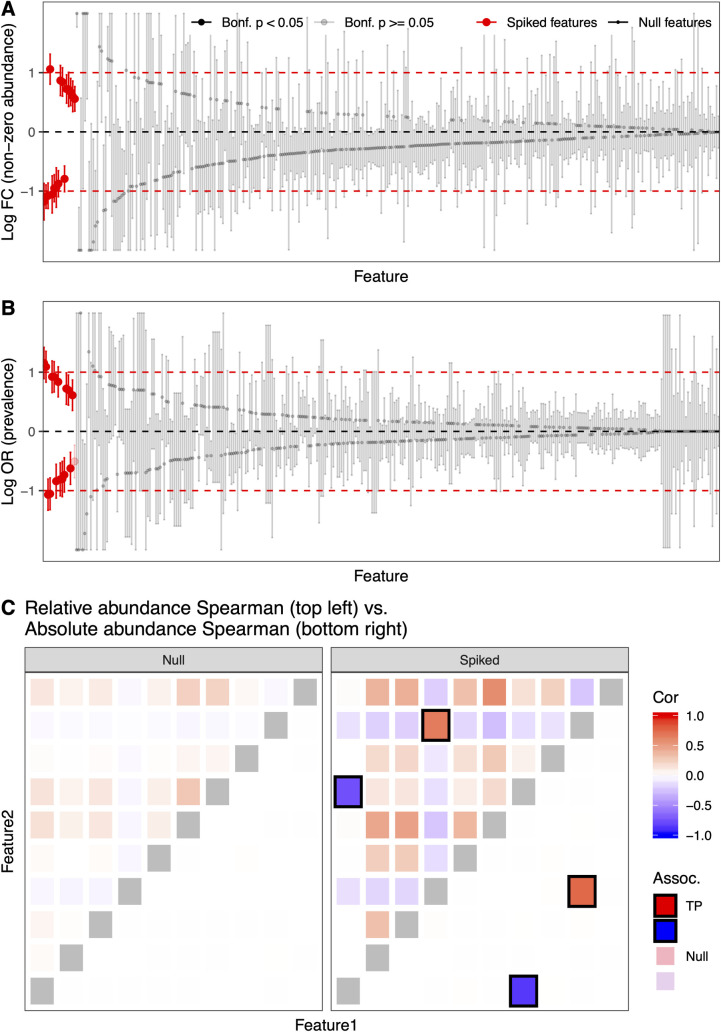
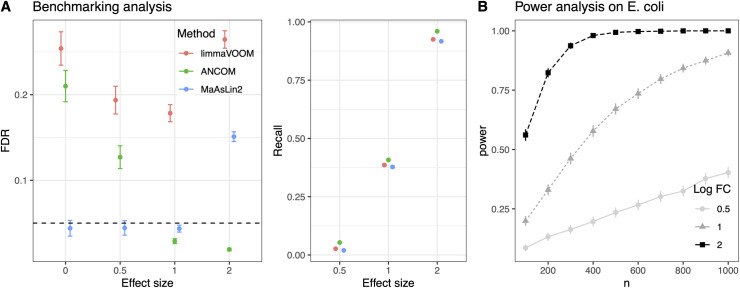
Many methods have been developed for statistical analysis of microbial community profiles, but due to the complex nature of typical microbiome measurements (e.g. sparsity, zero-inflation, non-independence, and compositionality) and of the associated underlying biology, it is difficult to compare or evaluate such methods within a single systematic framework. To address this challenge, we developed SparseDOSSA (Sparse Data Observations for the Simulation of Synthetic Abundances): a statistical model of microbial ecological population structure, which can be used to parameterize real-world microbial community profiles and to simulate new, realistic profiles of known structure for methods evaluation. Specifically, SparseDOSSA's model captures marginal microbial feature abundances as a zero-inflated log-normal distribution, with additional model components for absolute cell counts and the sequence read generation process, microbe-microbe, and microbe-environment interactions. Together, these allow fully known covariance structure between synthetic features (i.e. "taxa") or between features and "phenotypes" to be simulated for method benchmarking. Here, we demonstrate SparseDOSSA's performance for 1) accurately modeling human-associated microbial population profiles; 2) generating synthetic communities with controlled population and ecological structures; 3) spiking-in true positive synthetic associations to benchmark analysis methods; and 4) recapitulating an end-to-end mouse microbiome feeding experiment. Together, these represent the most common analysis types in assessment of real microbial community environmental and epidemiological statistics, thus demonstrating SparseDOSSA's utility as a general-purpose aid for modeling communities and evaluating quantitative methods. An open-source implementation is available at http://huttenhower.sph.harvard.edu/sparsedossa2.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
