

Rare genetic variability in human drug target genes modulates drug response and can guide precision medicine
- PMID: 34516913
- PMCID: PMC8442892
- DOI: 10.1126/sciadv.abi6856
Rare genetic variability in human drug target genes modulates drug response and can guide precision medicine
Abstract
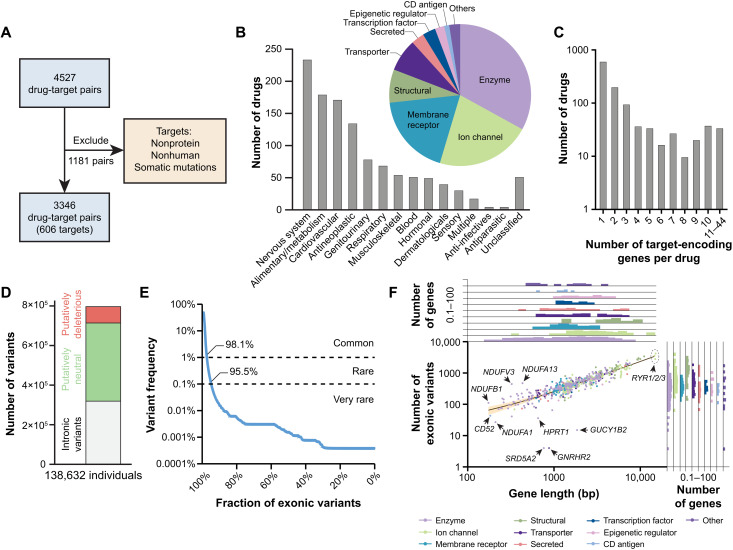
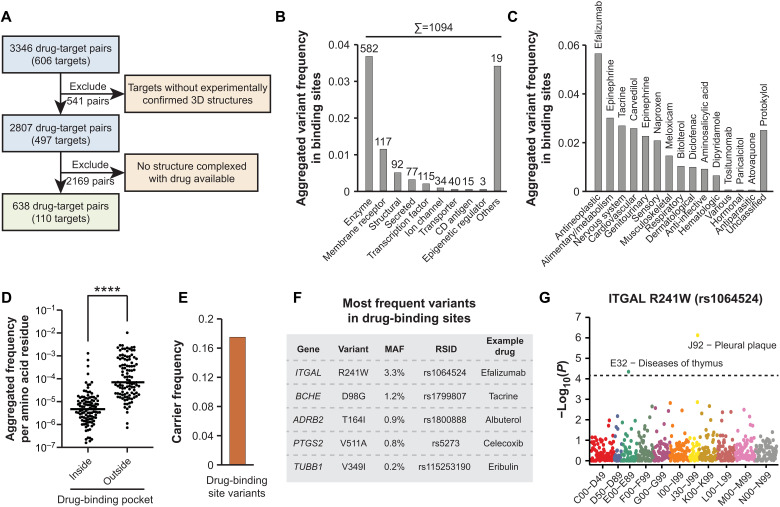
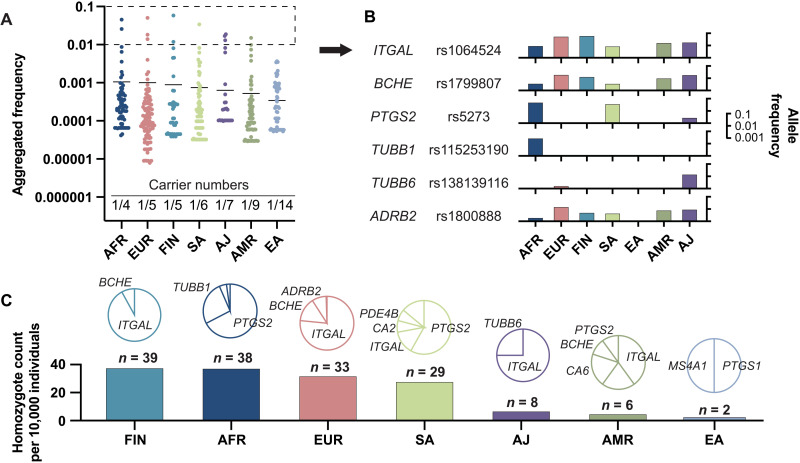
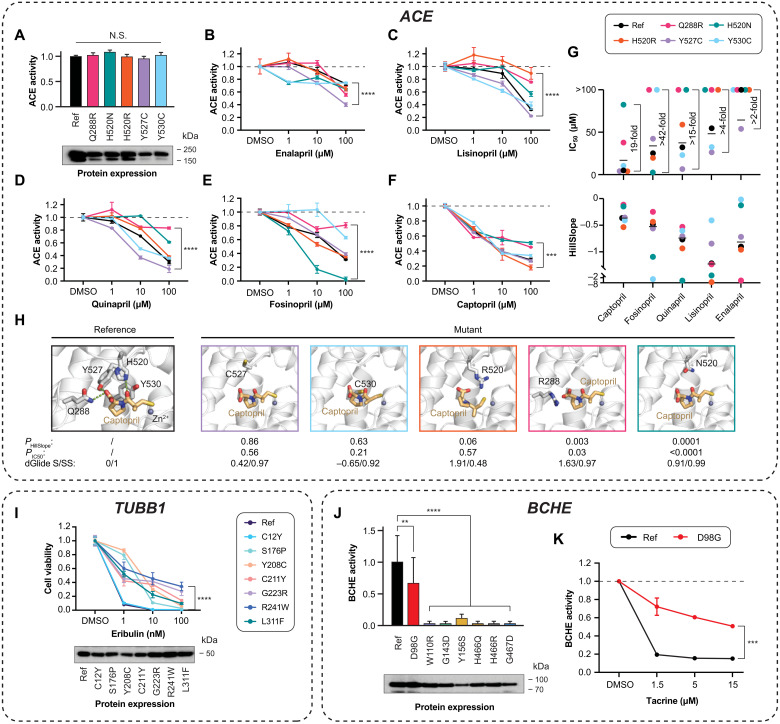
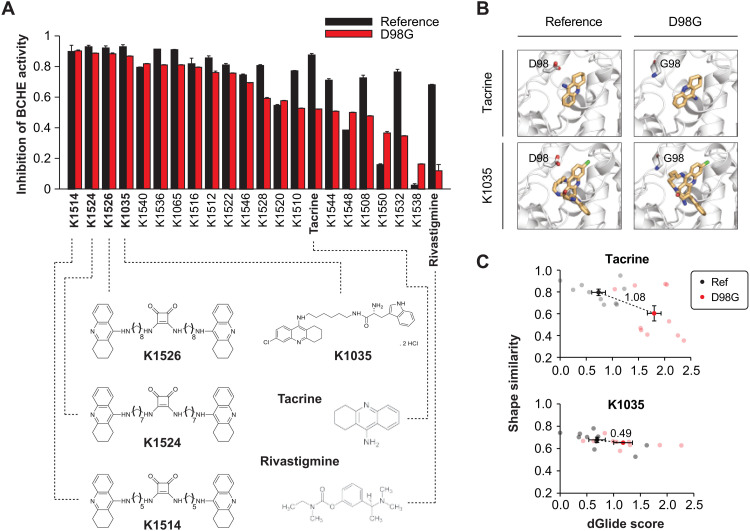
Interindividual variability in drug response constitutes a major concern in pharmacotherapy. While polymorphisms in genes involved in drug disposition have been extensively studied, drug target variability remains underappreciated. By mapping the genomic variability of all human drug target genes onto high-resolution crystal structures of drug target complexes, we identified 1094 variants localized within 6 Å of drug-binding pockets and directly affecting their geometry, topology, or physicochemical properties. We experimentally show that binding site variants affect pharmacodynamics with marked drug- and variant-specific differences. In addition, we demonstrate that a common BCHE variant confers resistance to tacrine and rivastigmine, which can be overcome by the use of derivatives based on squaric acid scaffolds or tryptophan conjugation. These findings underscore the importance of genetic drug target variability and demonstrate that integration of genomic data and structural information can inform personalized drug selection and genetically guided drug development to overcome resistance.
Figures
References
-
- Harrison R. K., Phase II and phase III failures: 2013–2015. Nat. Rev. Drug Discov. 15, 817–818 (2016). - PubMed
-
- Smietana K., Siatkowski M., Møller M., Trends in clinical success rates. Nat. Rev. Drug Discov. 15, 379–380 (2016). - PubMed
-
- Lacombe P. S., Vicente L., Pagès J. C., Morselli P. L., Causes and problems of nonresponse or poor response to drugs. Drugs 51, 552–570 (1996). - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
