

Multi-platform profiling characterizes molecular subgroups and resistance networks in chronic lymphocytic leukemia
- PMID: 34518531
- PMCID: PMC8438057
- DOI: 10.1038/s41467-021-25403-y
Multi-platform profiling characterizes molecular subgroups and resistance networks in chronic lymphocytic leukemia
Abstract
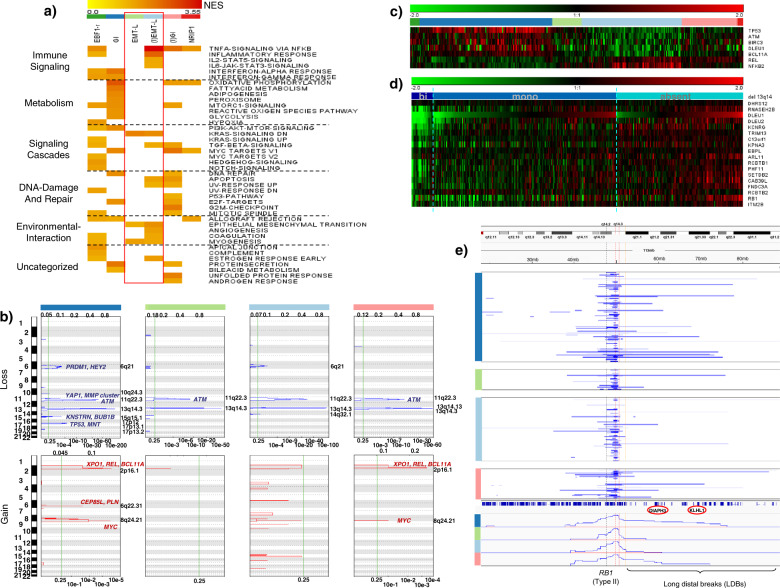
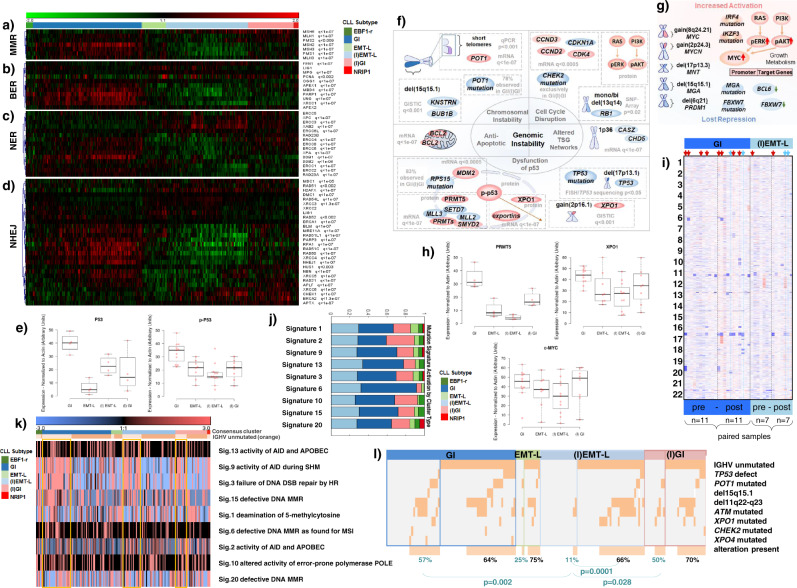
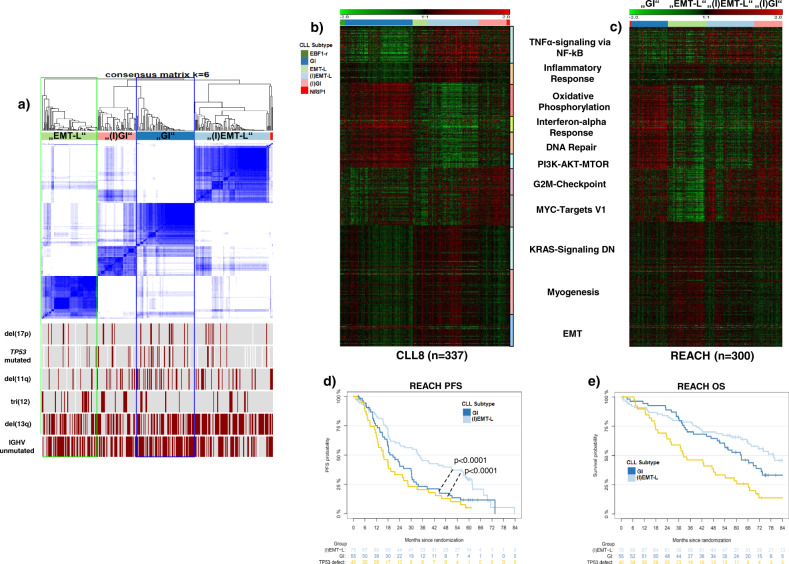
Knowledge of the genomic landscape of chronic lymphocytic leukemia (CLL) grows increasingly detailed, providing challenges in contextualizing the accumulated information. To define the underlying networks, we here perform a multi-platform molecular characterization. We identify major subgroups characterized by genomic instability (GI) or activation of epithelial-mesenchymal-transition (EMT)-like programs, which subdivide into non-inflammatory and inflammatory subtypes. GI CLL exhibit disruption of genome integrity, DNA-damage response and are associated with mutagenesis mediated through activation-induced cytidine deaminase or defective mismatch repair. TP53 wild-type and mutated/deleted cases constitute a transcriptionally uniform entity in GI CLL and show similarly poor progression-free survival at relapse. EMT-like CLL exhibit high genomic stability, reduced benefit from the addition of rituximab and EMT-like differentiation is inhibited by induction of DNA damage. This work extends the perspective on CLL biology and risk categories in TP53 wild-type CLL. Furthermore, molecular targets identified within each subgroup provide opportunities for new treatment approaches.
© 2021. The Author(s).
Conflict of interest statement
S.S. received advisory board honoraria, research support, travel support, speaker fees from AbbVie, Amgen, AstraZeneca, Celgene, Gilead, GSK, Hoffmann-La Roche, Janssen, Novartis, Sunesis. O.E. is supported by Janssen, Johnson and Johnson, Volastra Therapeutics, AstraZeneca, and Eli Lilly research grants. He is a scientific advisor and equity holder in Freenome, Owkin, Volastra Therapeutics and One Three Biotech and paid scientific advisor to Champion Oncology. M.S.C. is a retained consultant for BioInvent International and has performed educational and advisory roles for Roche, Boehringer Ingelheim, Baxalta, Merck KGaA, and GLG. He has received research funding from Bioinvent, Roche, Gilead, iTeos, UCB, and GSK. He is co-inventor of patent WO2012022985A1 protecting antibodies directed to hFcgRIIB in combination with CD20 specific antibodies. L.B.: Advisory Committees Abbvie, Amgen, Astellas, Bristol-Myers Squibb, Celgene, Daiichi Sankyo, Gilead, Hexal, Janssen, Jazz Pharmaceuticals, Menarini, Novartis, Pfizer, Sanofi, Seattle Genetics. R-F.Y. and M.W. are employed by Genentech and Roche, respectively. J.B. received travel support from Janssen and research support from Roche. The remaining authors declare no competing interests.
Figures




References
-
- Stankovic, T. et al. Ataxia telangiectasia mutated-deficient B-cell chronic lymphocytic leukemia occurs in pregerminal center cells and results in defective damage response and unrepaired chromosome damage. Blood10.1182/blood.V99.1.300 (2002). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
