

DNA methylation landscapes of 1538 breast cancers reveal a replication-linked clock, epigenomic instability and cis-regulation
- PMID: 34518533
- PMCID: PMC8437946
- DOI: 10.1038/s41467-021-25661-w
DNA methylation landscapes of 1538 breast cancers reveal a replication-linked clock, epigenomic instability and cis-regulation
Abstract
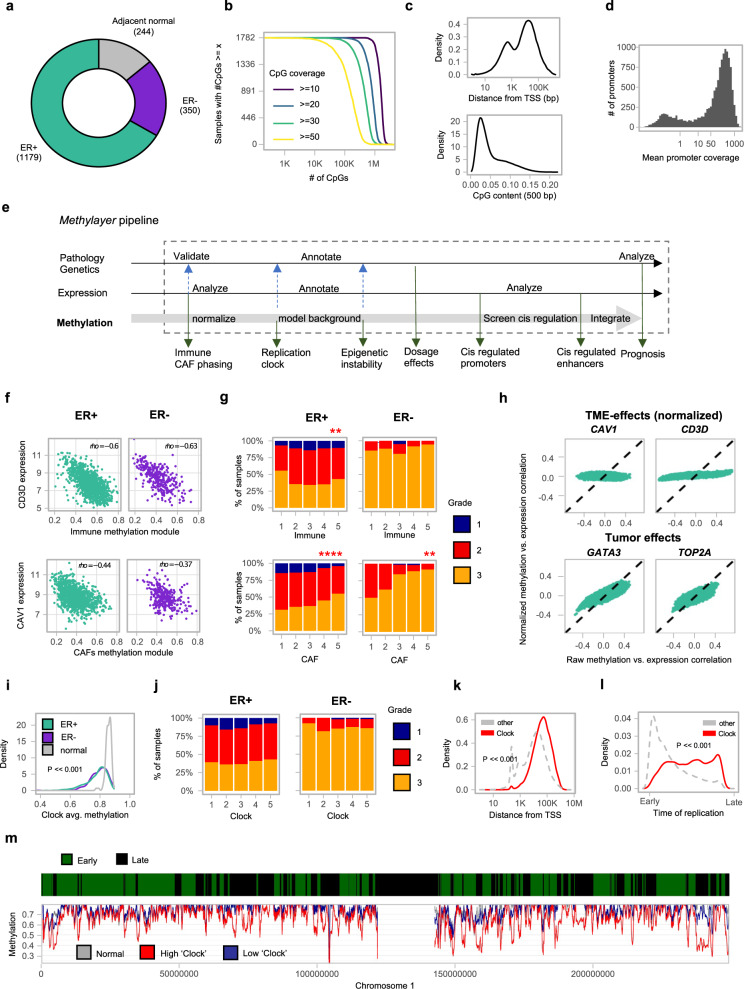
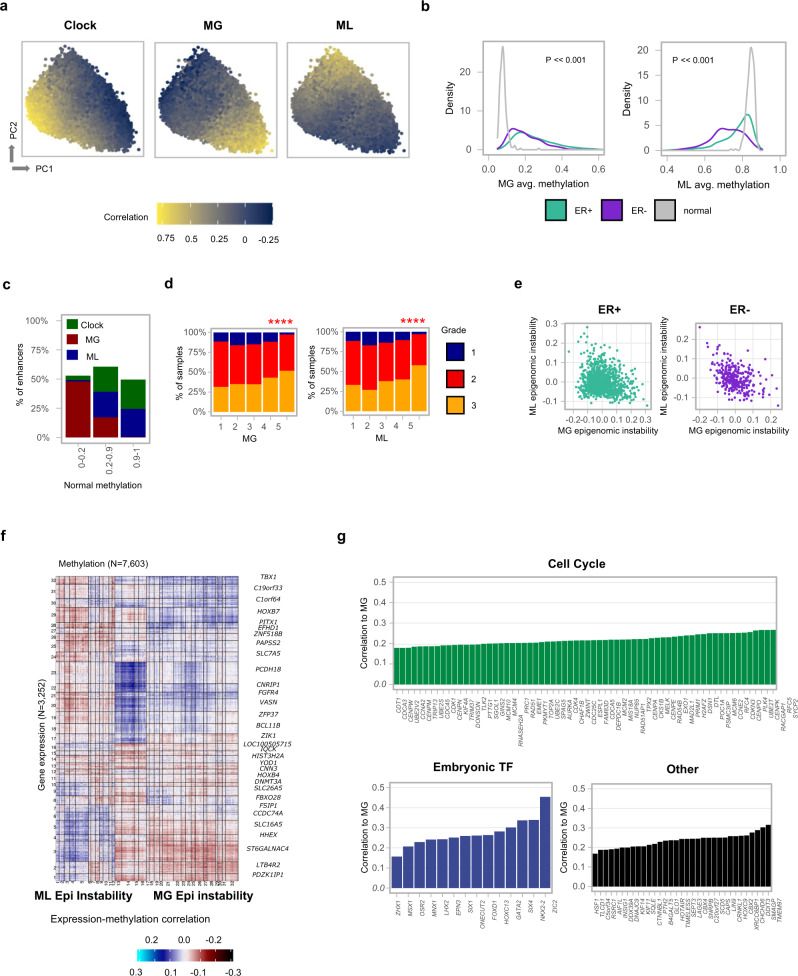
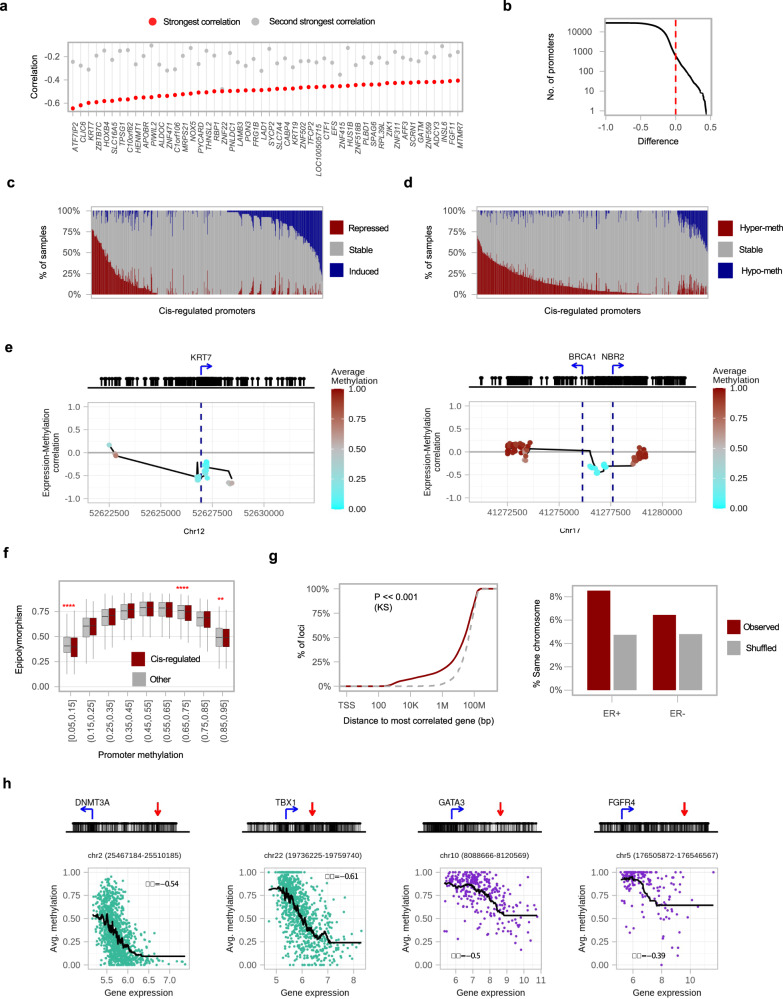
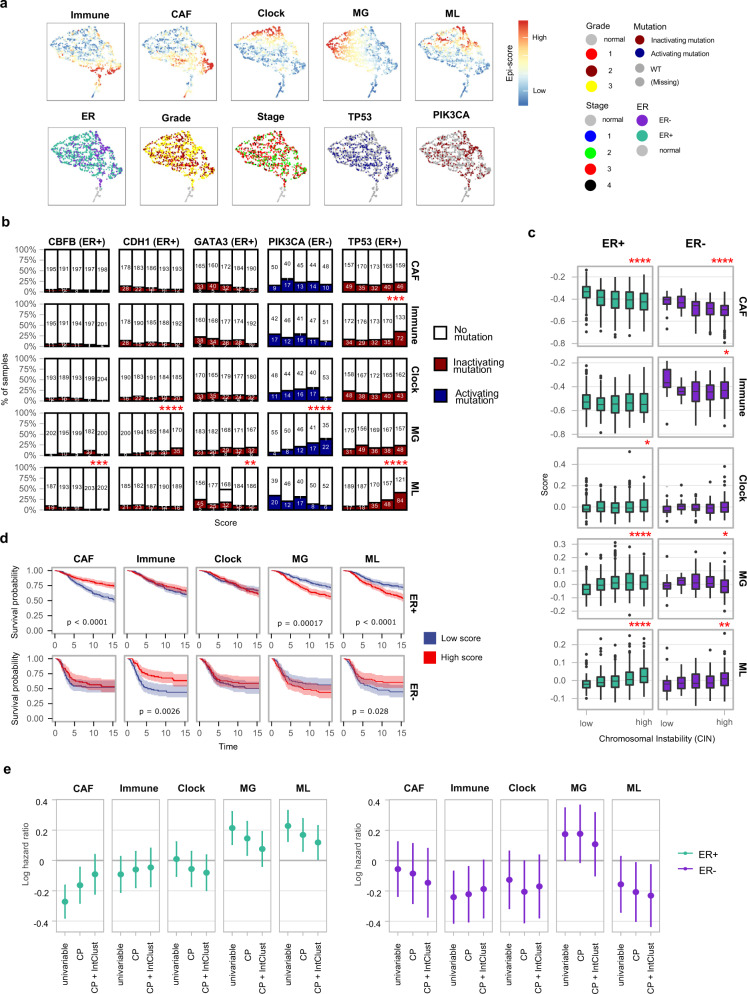
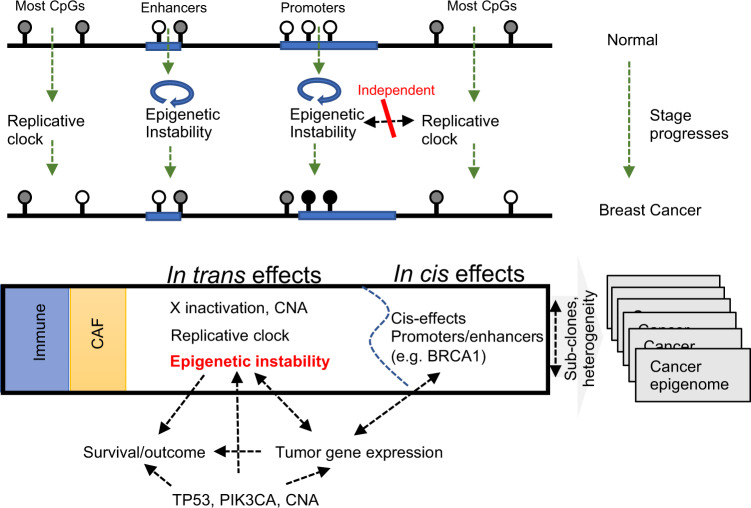
DNA methylation is aberrant in cancer, but the dynamics, regulatory role and clinical implications of such epigenetic changes are still poorly understood. Here, reduced representation bisulfite sequencing (RRBS) profiles of 1538 breast tumors and 244 normal breast tissues from the METABRIC cohort are reported, facilitating detailed analysis of DNA methylation within a rich context of genomic, transcriptional, and clinical data. Tumor methylation from immune and stromal signatures are deconvoluted leading to the discovery of a tumor replication-linked clock with genome-wide methylation loss in non-CpG island sites. Unexpectedly, methylation in most tumor CpG islands follows two replication-independent processes of gain (MG) or loss (ML) that we term epigenomic instability. Epigenomic instability is correlated with tumor grade and stage, TP53 mutations and poorer prognosis. After controlling for these global trans-acting trends, as well as for X-linked dosage compensation effects, cis-specific methylation and expression correlations are uncovered at hundreds of promoters and over a thousand distal elements. Some of these targeted known tumor suppressors and oncogenes. In conclusion, this study demonstrates that global epigenetic instability can erode cancer methylomes and expose them to localized methylation aberrations in-cis resulting in transcriptional changes seen in tumors.
© 2021. The Author(s).
Conflict of interest statement
S.A.J.R.A. is founder and shareholder of Contextual Genomic and a scientific advisor to Sangamo Biosciences and Takeda Pharmaceuticals. C.C. is a scientific advisor to AstraZeneca-iMed and has received research funding (administered by the University of Cambridge) from AstraZeneca, Servier, and Genentech/Roche.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
