

This is a preprint.
Rescuing Low Frequency Variants within Intra-Host Viral Populations directly from Oxford Nanopore sequencing data
- PMID: 34518837
- PMCID: PMC8437309
- DOI: 10.1101/2021.09.03.458038
Rescuing Low Frequency Variants within Intra-Host Viral Populations directly from Oxford Nanopore sequencing data
Update in
-
Rescuing low frequency variants within intra-host viral populations directly from Oxford Nanopore sequencing data.Nat Commun. 2022 Mar 14;13(1):1321. doi: 10.1038/s41467-022-28852-1. Nat Commun. 2022. PMID: 35288552 Free PMC article.
Abstract
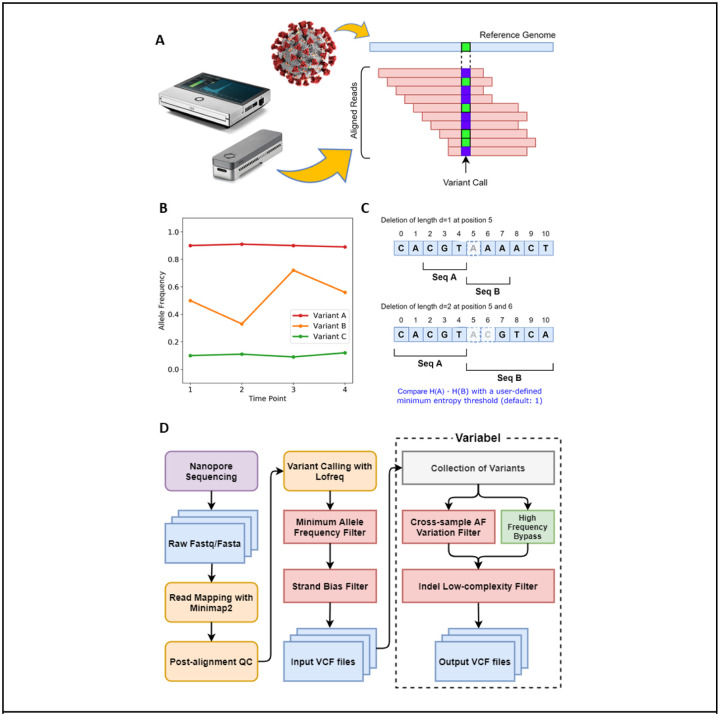
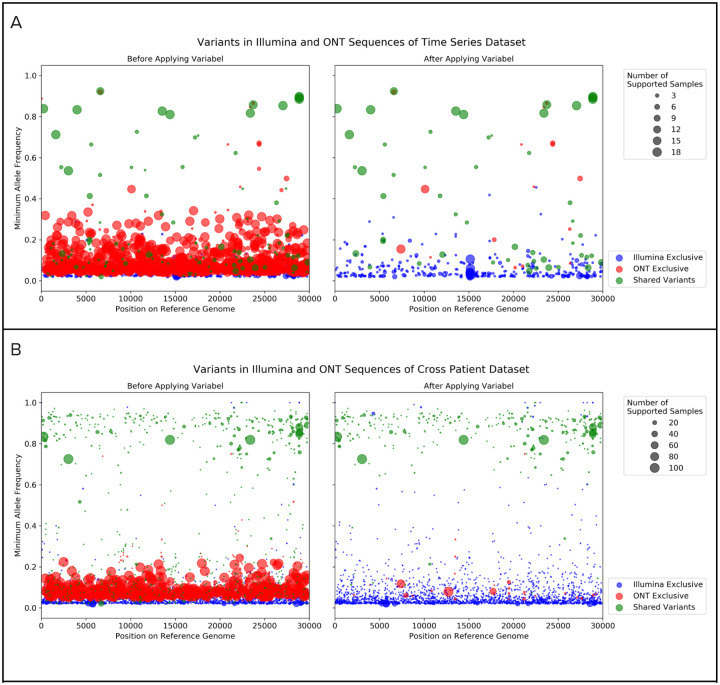
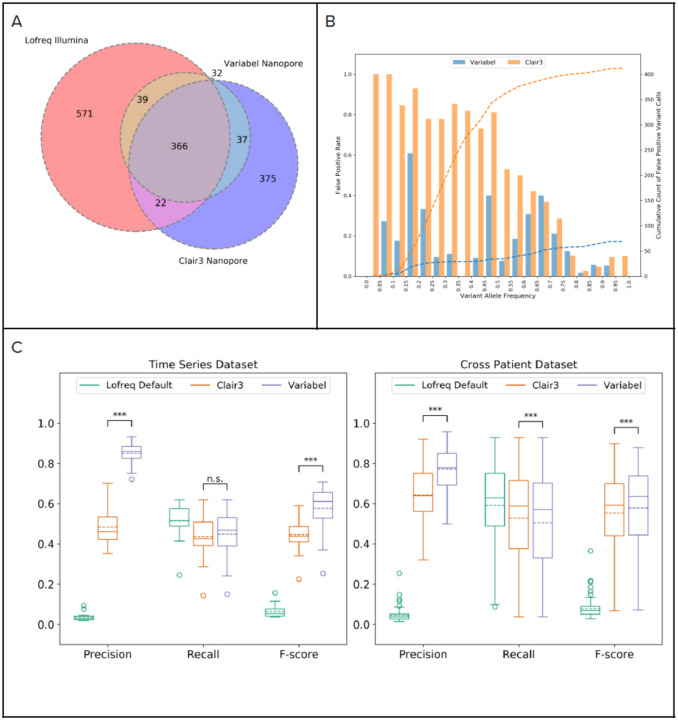
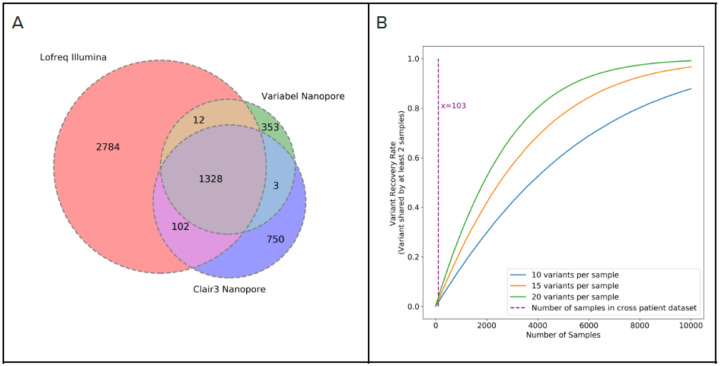
Infectious disease monitoring on Oxford Nanopore Technologies (ONT) platforms offers rapid turnaround times and low cost, exemplified by well over a half of million ONT SARS-COV-2 datasets. Tracking low frequency intra-host variants has provided important insights with respect to elucidating within host viral population dynamics and transmission. However, given the higher error rate of ONT, accurate identification of intra-host variants with low allele frequencies remains an open challenge with no viable solutions available. In response to this need, we present Variabel, a novel approach and first method designed for rescuing low frequency intra-host variants from ONT data alone. We evaluated Variabel on both within patient and across patient paired Illumina and ONT datasets; our results show that Variabel can accurately identify low frequency variants below 0.5 allele frequency, outperforming existing state-of-the-art ONT variant callers for this task. Variabel is open-source and available for download at: www.gitlab.com/treangenlab/variabel.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous