

SARS-CoV-2 structural coverage map reveals viral protein assembly, mimicry, and hijacking mechanisms
- PMID: 34519429
- PMCID: PMC8438690
- DOI: 10.15252/msb.202010079
SARS-CoV-2 structural coverage map reveals viral protein assembly, mimicry, and hijacking mechanisms
Abstract
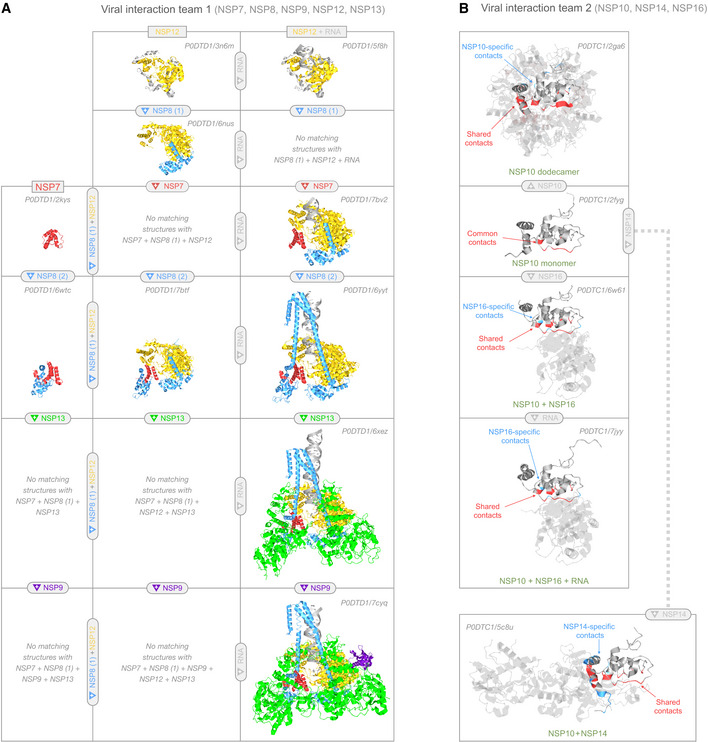
We modeled 3D structures of all SARS-CoV-2 proteins, generating 2,060 models that span 69% of the viral proteome and provide details not available elsewhere. We found that ˜6% of the proteome mimicked human proteins, while ˜7% was implicated in hijacking mechanisms that reverse post-translational modifications, block host translation, and disable host defenses; a further ˜29% self-assembled into heteromeric states that provided insight into how the viral replication and translation complex forms. To make these 3D models more accessible, we devised a structural coverage map, a novel visualization method to show what is-and is not-known about the 3D structure of the viral proteome. We integrated the coverage map into an accompanying online resource (https://aquaria.ws/covid) that can be used to find and explore models corresponding to the 79 structural states identified in this work. The resulting Aquaria-COVID resource helps scientists use emerging structural data to understand the mechanisms underlying coronavirus infection and draws attention to the 31% of the viral proteome that remains structurally unknown or dark.
Keywords: COVID-19; SARS-CoV-2; bioinformatics; data visualization; structural biology.
© 2021 The Authors. Published under the terms of the CC BY 4.0 license.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
- A
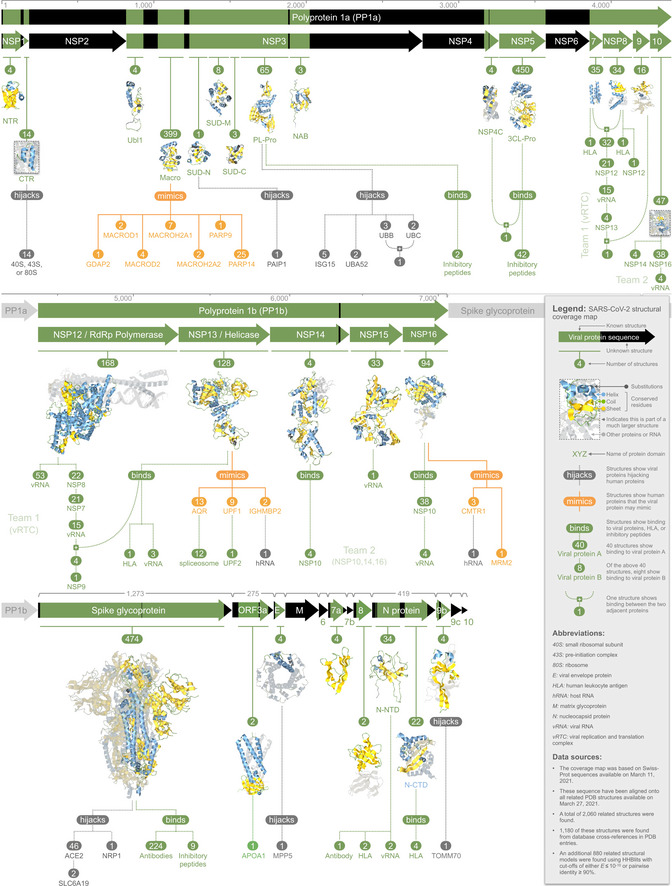
Lists domain topology for seven human proteins potentially mimicked by the macro domain of NSP3. The list was ranked by alignment significance (HHblits E‐value) and includes a summary of potentially mimicked functions. Each macro domain is numbered to indicate its CATH functional family. The top‐ranked proteins (MACROD2 and MACROD1) remove ADPr from proteins, reversing the effect of ADPr writers (PARP14 and PARP9), and affecting ADPr readers (GDAP2, MACROH2A1, and MACROH2A2). For PARP9 and PARP14, the table indicates the best alignment of the NSP3 sequence onto the available structures corresponding to each macro domain.
- B
Lists three human helicase proteins potentially mimicked by NSP13. The list was ranked by alignment significance (HHblits E‐value) and includes a summary of potentially mimicked functions. We found stronger evidence for mimicry by NSP13 than by NSP3. For each human protein, the 3D structure is shown with Aquaria’s default coloring scheme, in this case indicating the region of alignment with NSP13 (Fig 1, Dataset EV4). For UPF1 (
https://aquaria.ws/P0DTD1/2wjv ), the structure coloring reveals that UPF2 binds to a region not matched by NSP13, suggesting that NSP13 may not bind UPF2. For IGHMBP2 (https://aquaria.ws/P0DTD1/4b3g ), the structure coloring reveals that RNA binds to the region matched by NSP13, suggesting that NSP13 binds RNA. For AQR (https://aquaria.ws/P0DTD1/6jyt ), the structure coloring reveals that the spliceosome binds to a region not matched by NSP13, suggesting that NSP13 may not bind the spliceosome.
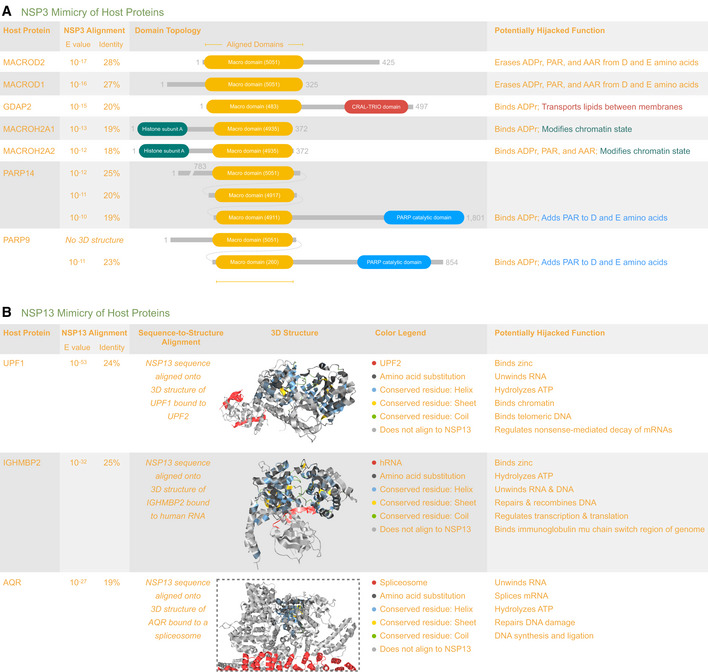
- A
Hijacking of ribosomal complexes is shown in 14 matching structures, most of which were determined using the full‐length sequence of NSP1 (180 residues); however, only a ˜36 residue fragment was ordered enough to appear in the structures. The coloring scheme highlights the location of this fragment within the ribosome (
https://aquaria.ws/P0DTC1/6zlw ), revealing how NSP1 blocks host mRNA translation (Thoms et al, 2020). - B
Hijacking of PAIP1 (a.k.a. “PABP‐interacting protein 1”) is shown in only one matching structure that was determined using the SUD‐N region of NSP3 from SARS‐CoV (Nikulin et al, 2021). The structure (
https://aquaria.ws/P0DTC1/6yxj ) shows the strong overall sequence similarity in SARS‐CoV‐2 and reveals that, of the 15 residues contacting PAIP1, 13 are identical in SARS‐CoV‐2. - C
Hijacking of ubiquitin‐like (Ubl) domains is shown in 10 matching structures, of which only one showed simultaneous binding to two Ubl domains (shown above). The structure (
https://aquaria.ws/P0DTC1/5e6j ) was determined using NSP3 from SARS‐CoV (Békés et al, 2016), which had strong overall sequence similarity in SARS‐CoV‐2; of the 31 residues contacting UBB or UBC, 27 are identical in SARS‐COV‐2. - D
Hijacking of ACE2 is shown in 46 matching structures; however, only two also show binding to SLC6A19 (Yan et al, 2020). In the structure shown here (
https://aquaria.ws/P0DTC2/6m17 ), spike glycoprotein does not directly bind to SLC6A19. - E
Hijacking of NRP1 (a.k.a. neuropilin‐1) is shown in only one matching structure (
https://aquaria.ws/P0DTC2/7jjc ), which includes only a three‐residue region from spike glycoprotein (Daly et al, 2020). - F
Hijacking of MPP5 (a.k.a. PALS1, “protein associated with Lin‐7 1”) is shown in only one matching structure (
https://aquaria.ws/P0DTC4/7m4r ), which includes only a nine‐residue region from envelope protein (Liu & Chai, 2021). - G
Hijacking of TOMM70 (a.k.a. “translocase of outer mitochondrial membrane protein 70”) is shown in only one matching structure (
https://aquaria.ws/P0DTD2/7kdt ), which includes only a 38‐residue region from ORF9b protein (Gordon et al, 2020).
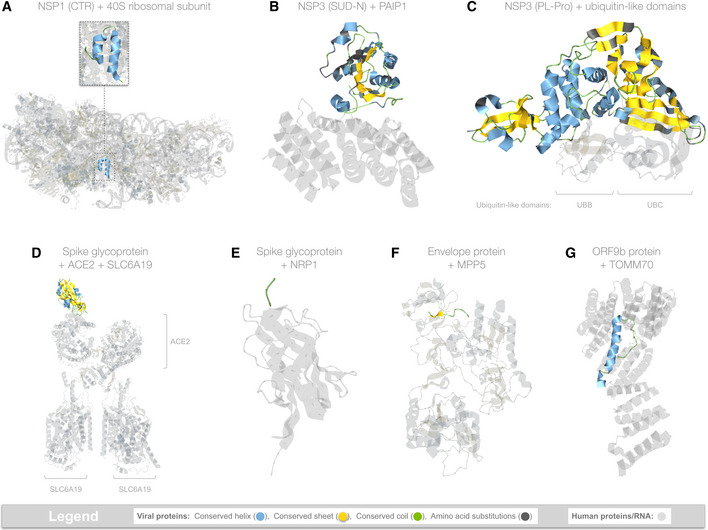
- A
In team 1, NSP7 (red), NSP8 (cyan), NSP9 (purple), NSP12 (yellow), and NSP13 (green) assemble into the replication and translation complex (RTC). NSP12 alone (top row, left) can replicate RNA (top row, right). NSP8 binds NSP12 at two sites: (i) at the NSP12 core (2nd row, left); and (ii) via NSP7‐mediated cooperative interactions with NSP12 (4th row, center), greatly enhancing RNA replication (4th row, right). NSP7 + NSP8 alone form a dimer in most structures (4th row, left), but can also form a tetramer (e.g.,
https://aquaria.ws/P0DTD1/7jlt ) or hexadecamer (e.g.,https://aquaria.ws/P0DTD1/2ahm ). Replication is also enhanced by NSP13 (5th row, right) and NSP9 (bottom row, right). - B
In team 2, NSP10 monomers (2nd row) can either self‐assemble into a spherical dodecamer (top), dimerize with NSP14 (bottom row), or dimerize with NSP16 (third row). The NSP10 + NSP16 heterodimer was also seen bound to a three‐residue RNA segment (fourth row). Residue coloring is used to show that NSP10, NSP14, and NSP16 appear to interact competitively, as noted in previous studies. In the structures shown, nine NSP10 residues (shown in red on the monomer) formed common intermolecular contacts in all three oligomers. Within each oligomer, most NSP10 residues involved in intermolecular contacts were shared (red) with at least one other oligomer; very few NSP10 residues formed contacts specific to that oligomer (blue).
References
-
- Bailey‐Elkin BA, Knaap RCM, Johnson GG, Dalebout TJ, Ninaber DK, van Kasteren PB, Bredenbeek PJ, Snijder EJ, Kikkert M, Mark BL (2014) Crystal structure of the middle east respiratory syndrome coronavirus (MERS‐CoV) papain‐like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J Biol Chem 289: 34667–34682 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
