

The roles of epigenetics in cancer progression and metastasis
- PMID: 34520519
- PMCID: PMC8524384
- DOI: 10.1042/BCJ20210084
The roles of epigenetics in cancer progression and metastasis
Abstract
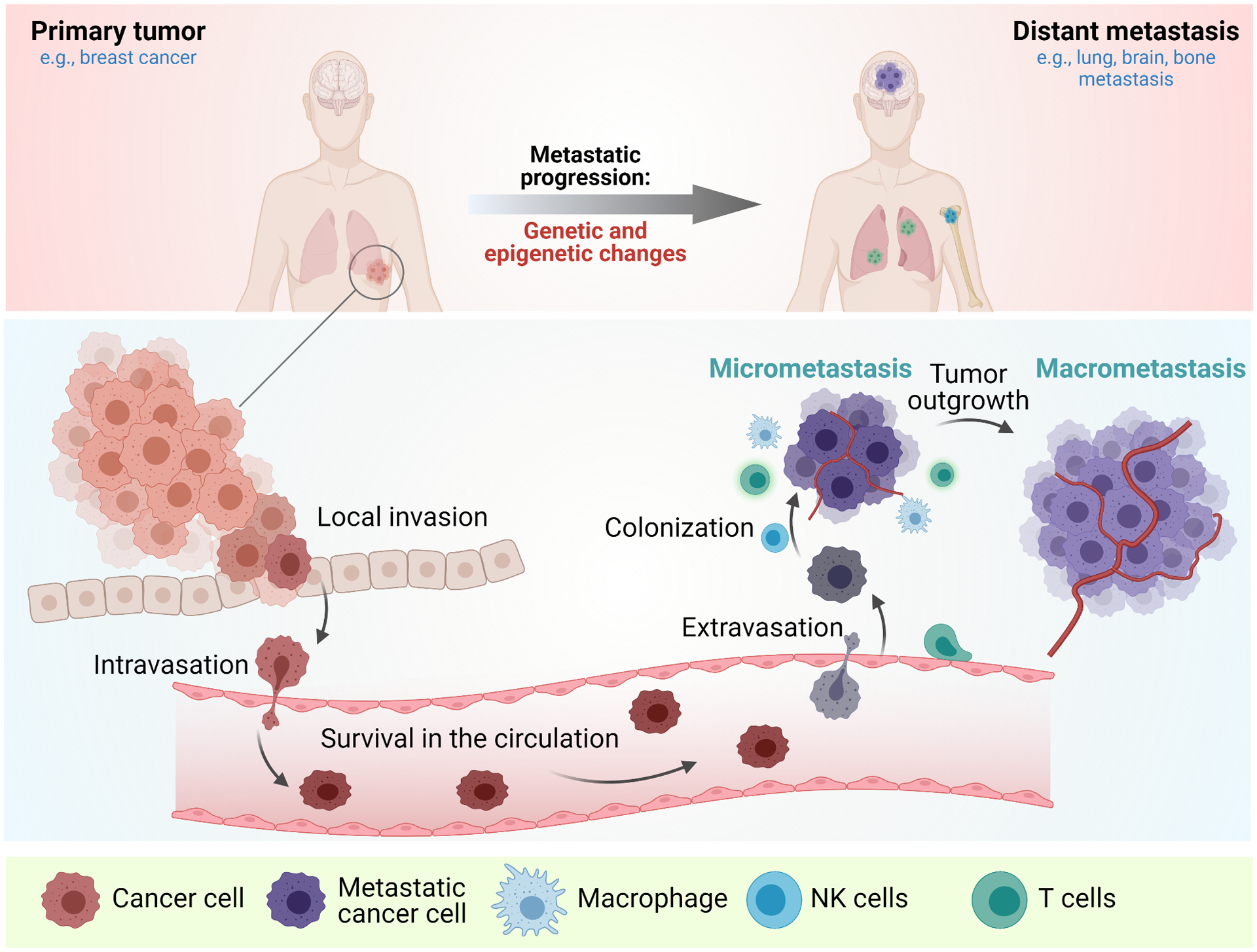
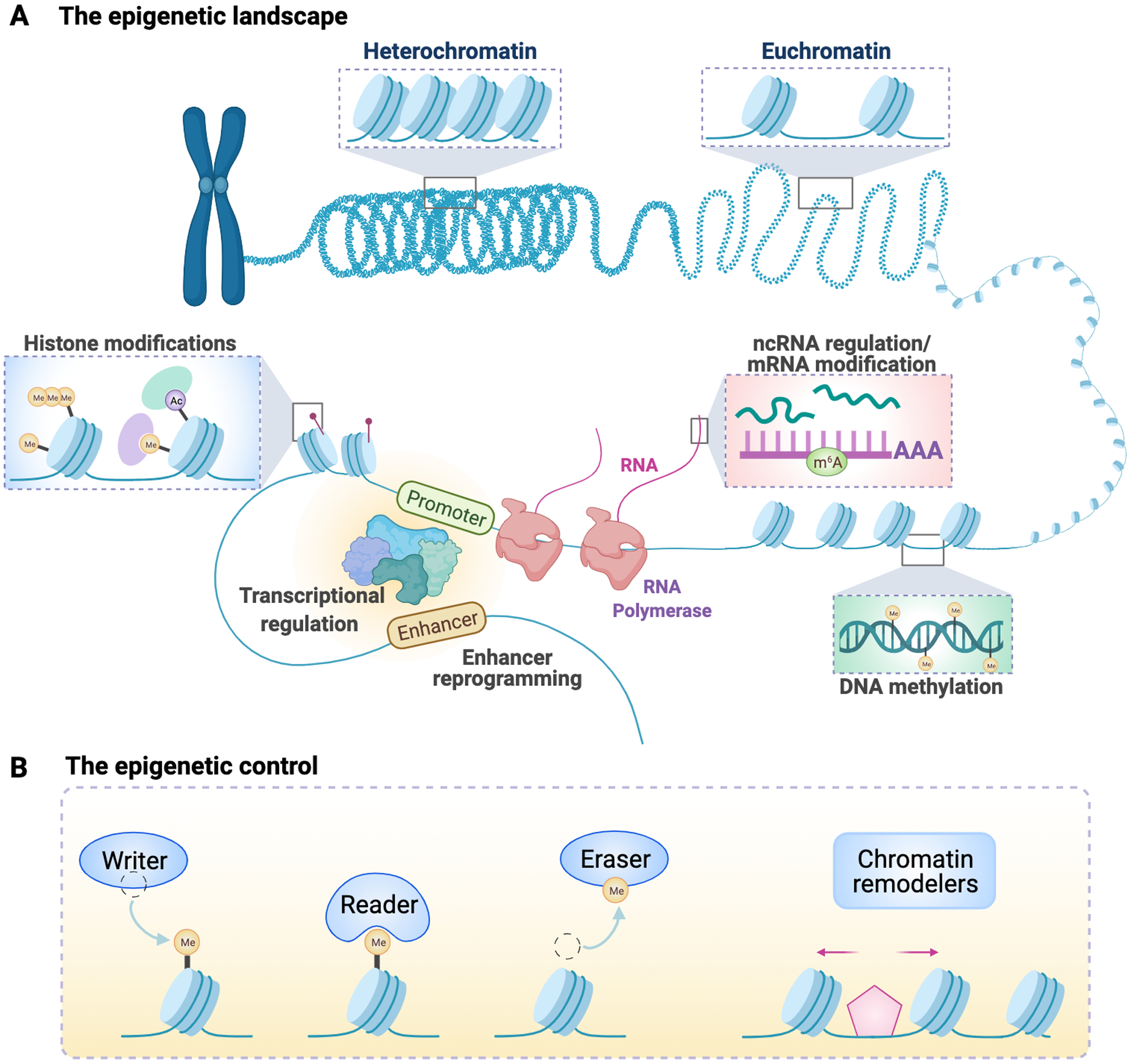
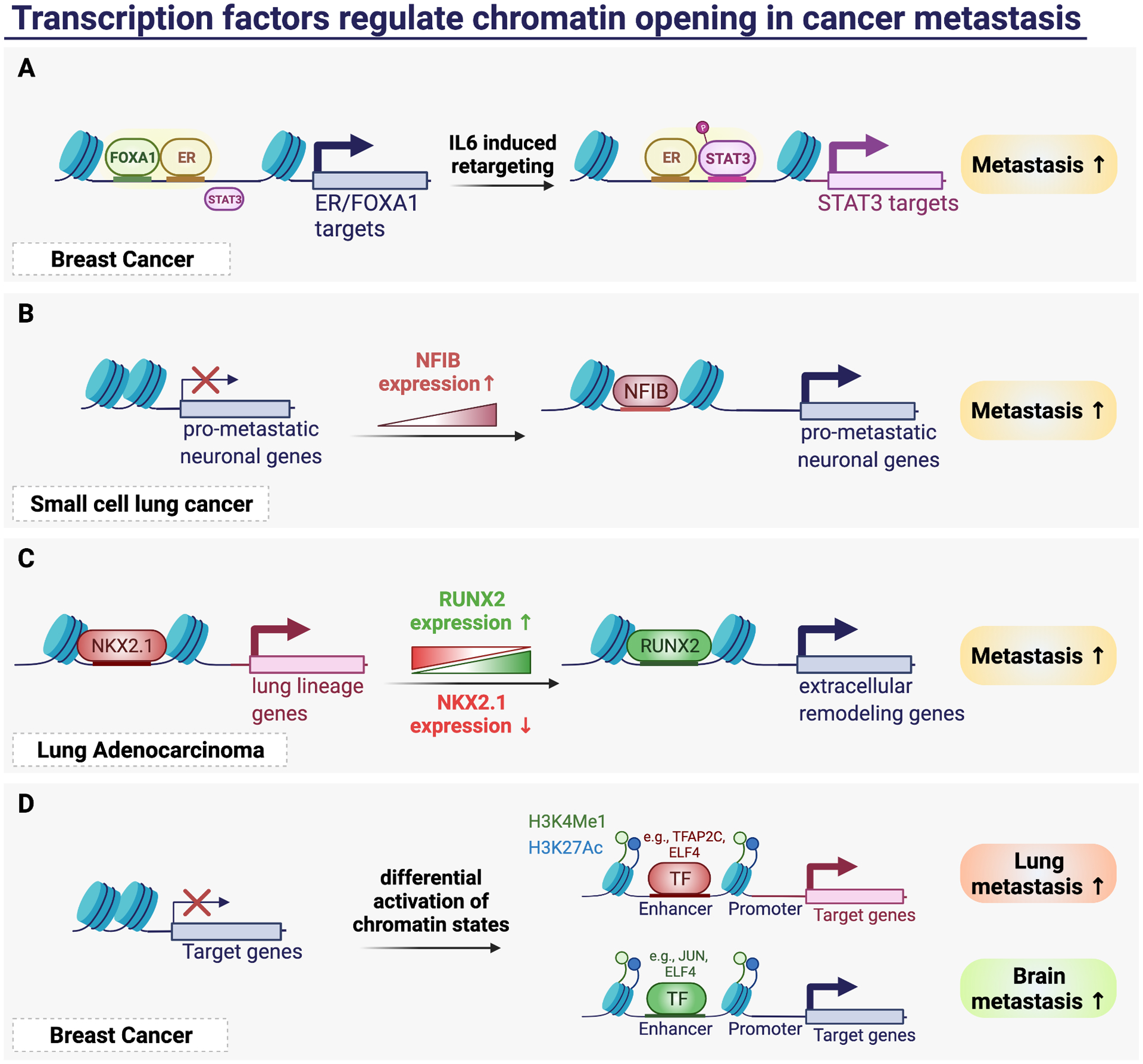
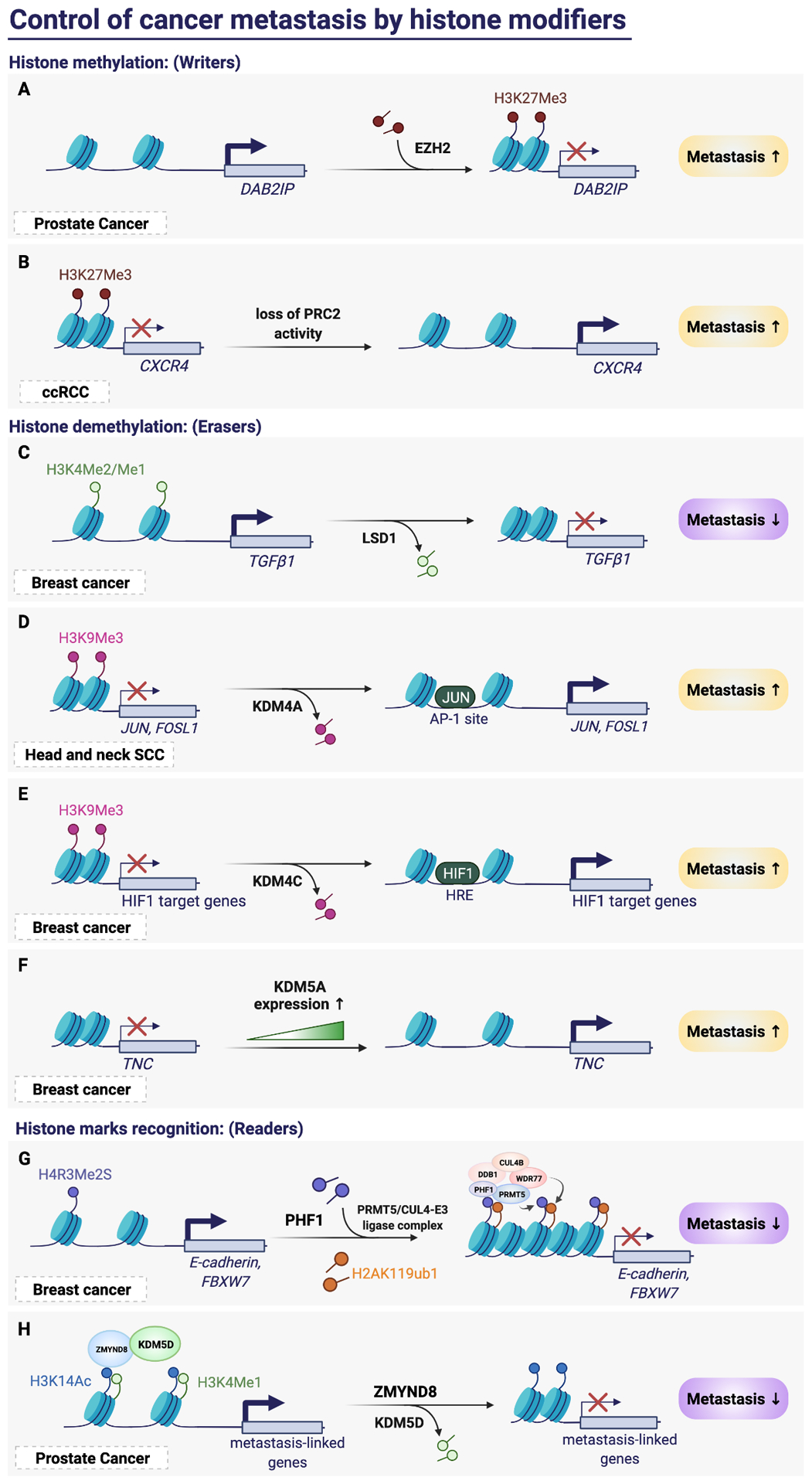
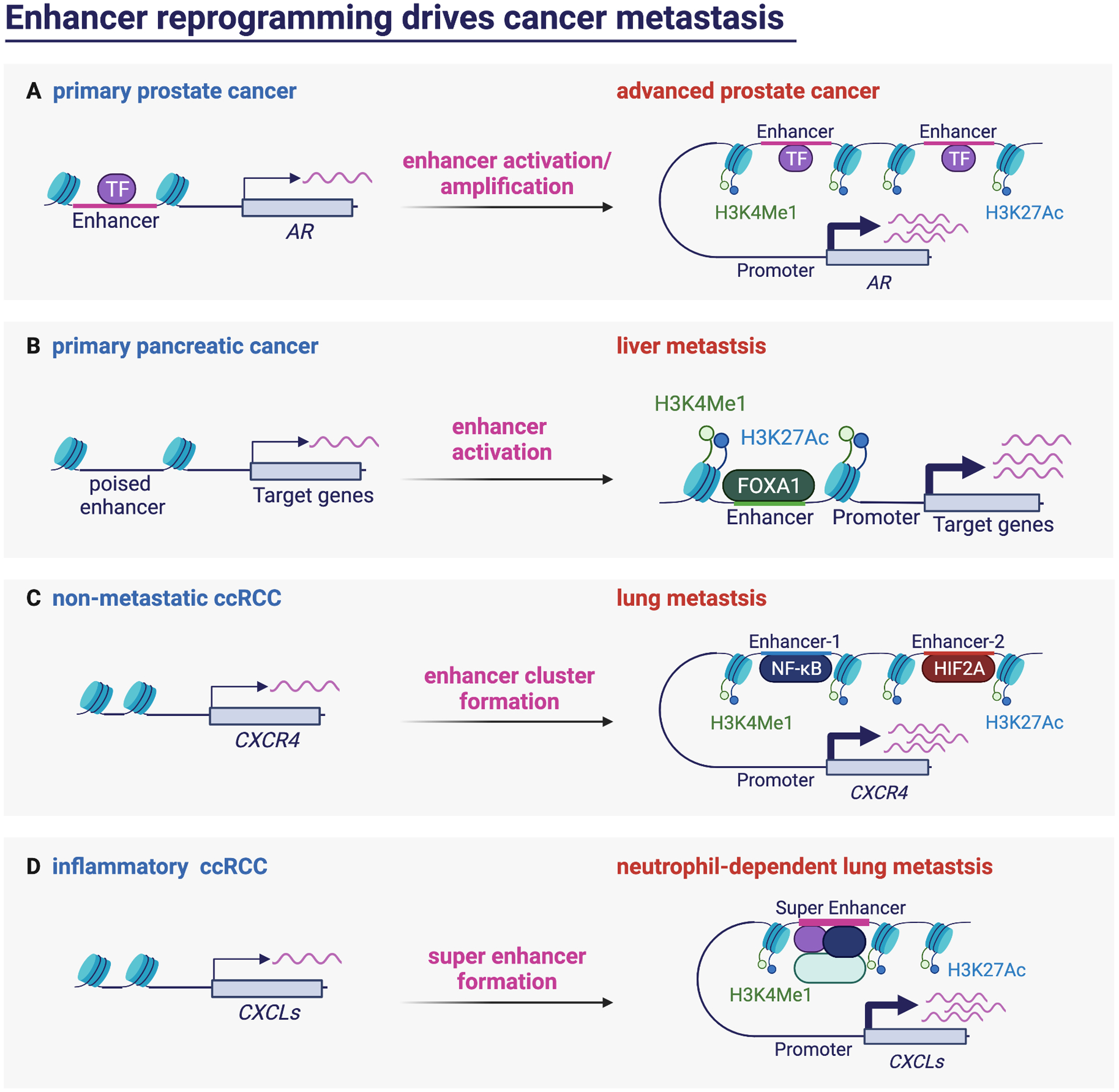
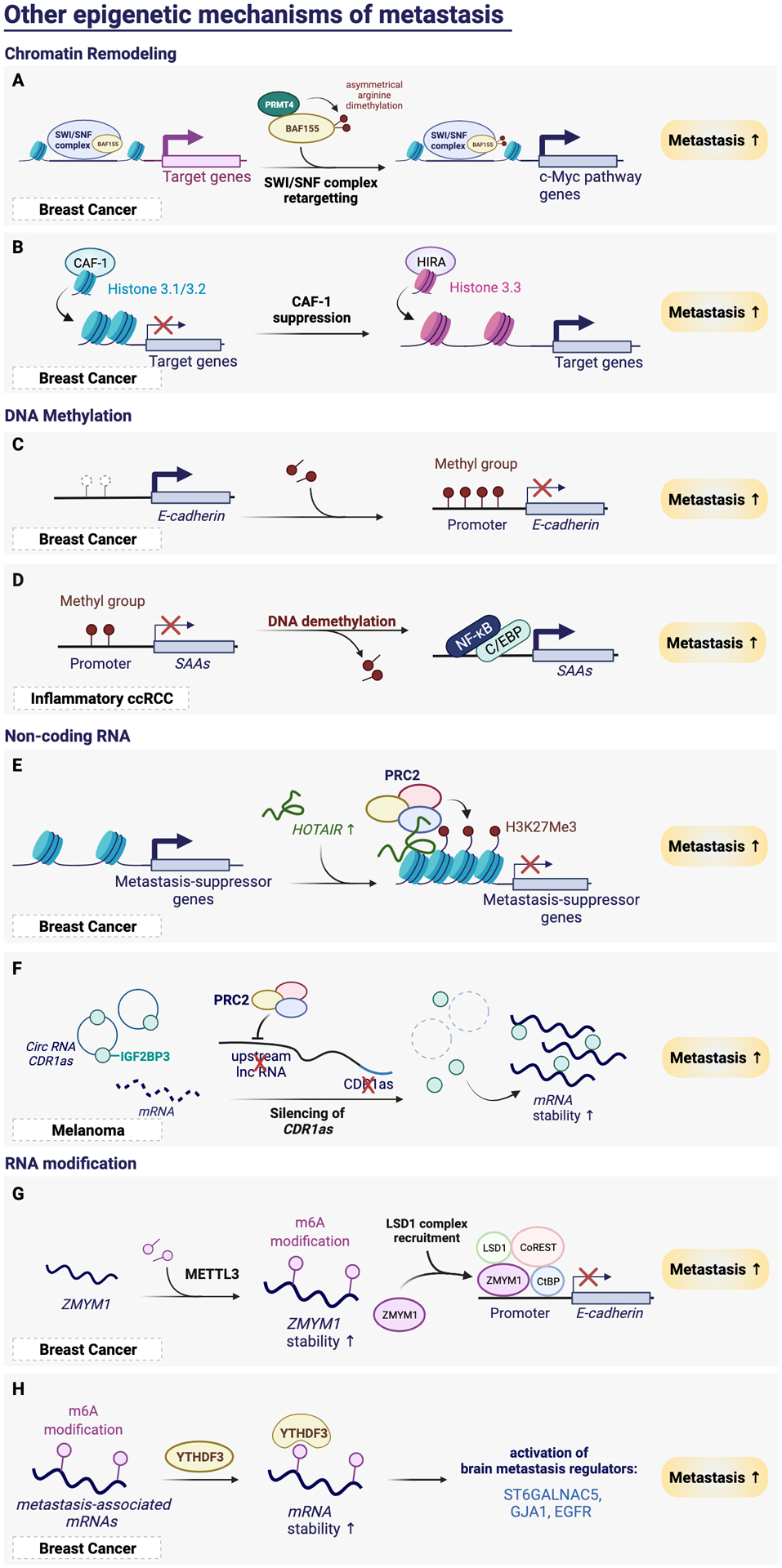
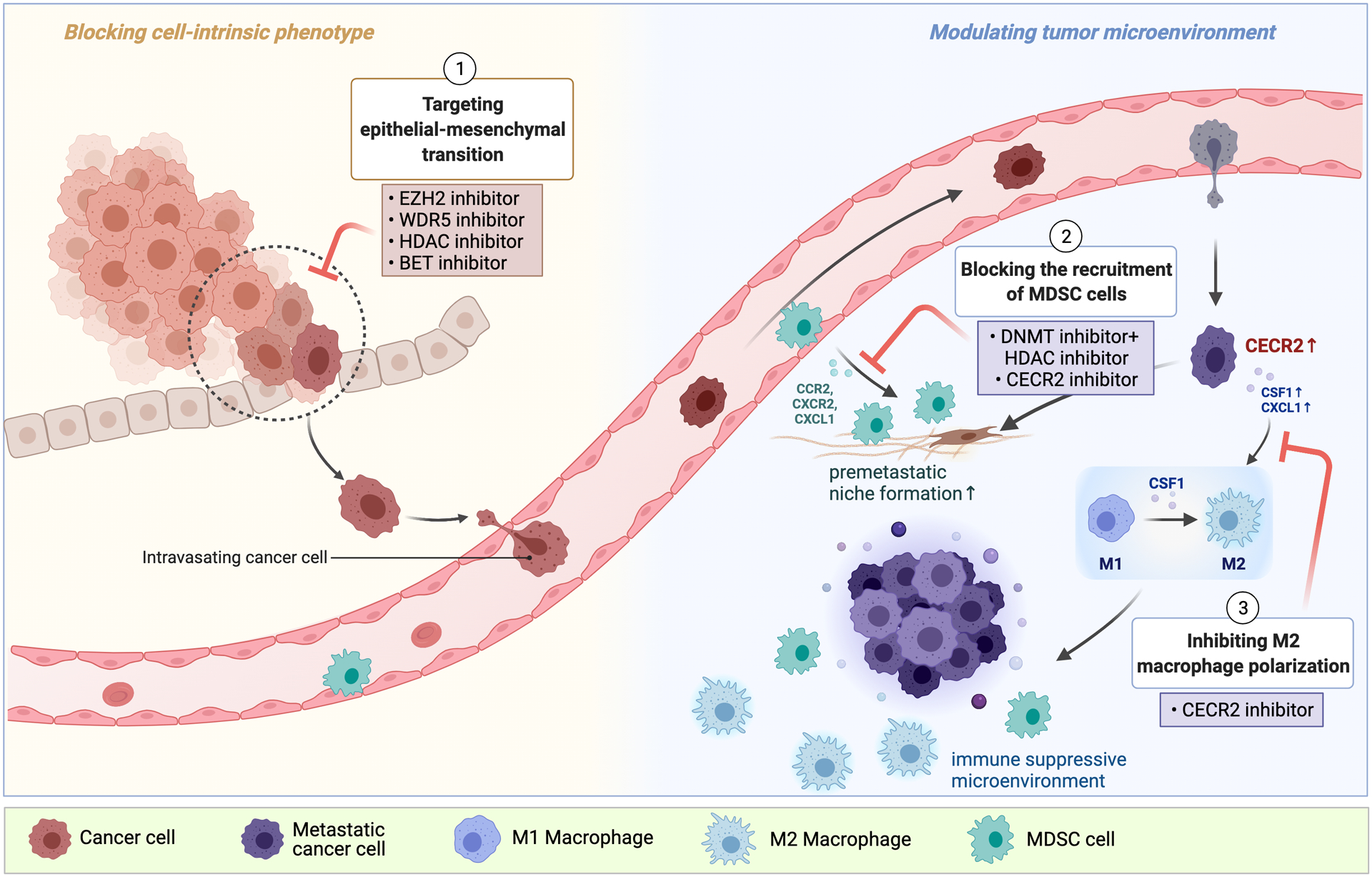
Cancer metastasis remains a major clinical challenge for cancer treatment. It is therefore crucial to understand how cancer cells establish and maintain their metastatic traits. However, metastasis-specific genetic mutations have not been identified in most exome or genome sequencing studies. Emerging evidence suggests that key steps of metastasis are controlled by reversible epigenetic mechanisms, which can be targeted to prevent and treat the metastatic disease. A variety of epigenetic mechanisms were identified to regulate metastasis, including the well-studied DNA methylation and histone modifications. In the past few years, large scale chromatin structure alterations including reprogramming of the enhancers and chromatin accessibility to the transcription factors were shown to be potential driving force of cancer metastasis. To dissect the molecular mechanisms and functional output of these epigenetic changes, it is critical to use advanced techniques and alternative animal models for interdisciplinary and translational research on this topic. Here we summarize our current understanding of epigenetic aberrations in cancer progression and metastasis, and their implications in developing new effective metastasis-specific therapies.
Keywords: cancer metastasis; chromatin opening; enhancer reprogramming; epigenetics; histone modification; tumor progression.
© 2021 The Author(s). Published by Portland Press Limited on behalf of the Biochemical Society.
Conflict of interest statement
Competing Interests
The authors declare no competing interests.
Figures
References
-
- Wittekind C, Neid M. Cancer invasion and metastasis. Oncology 2005;69 Suppl 1:14–6. - PubMed
-
- Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science (New York, NY) 2011;331:1559–64. - PubMed
-
- Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nature reviews Cancer 2009;9:274–84. - PubMed
-
- Fidler IJ. Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125 I-5-iodo-2’-deoxyuridine. Journal of the National Cancer Institute 1970;45:773–82. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous