

Pandora: nucleotide-resolution bacterial pan-genomics with reference graphs
- PMID: 34521456
- PMCID: PMC8442373
- DOI: 10.1186/s13059-021-02473-1
Pandora: nucleotide-resolution bacterial pan-genomics with reference graphs
Abstract
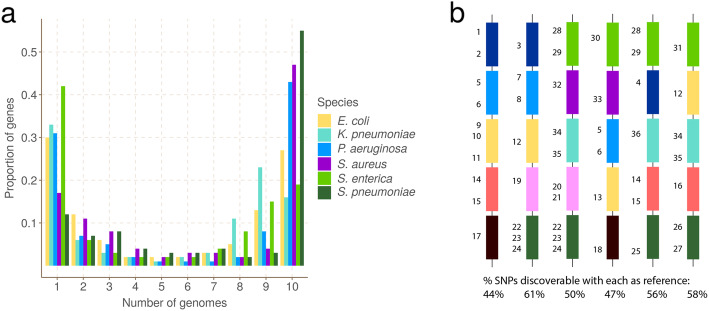
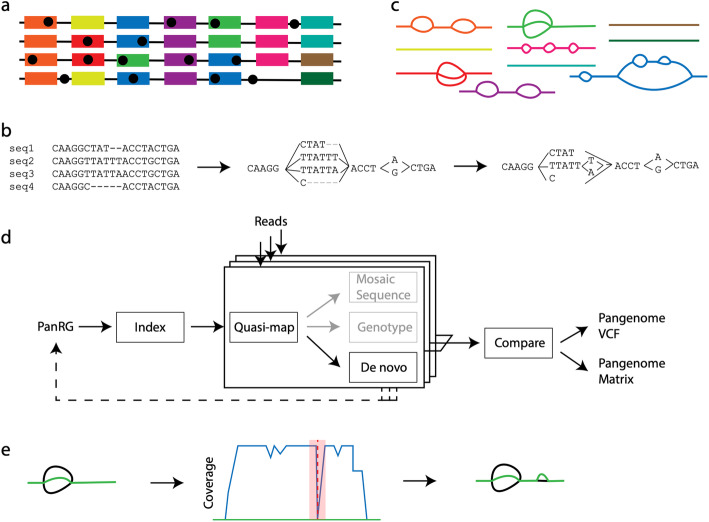
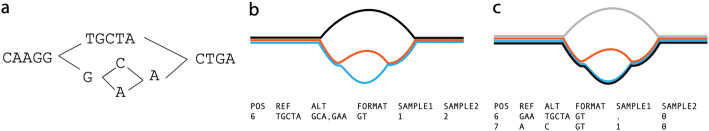
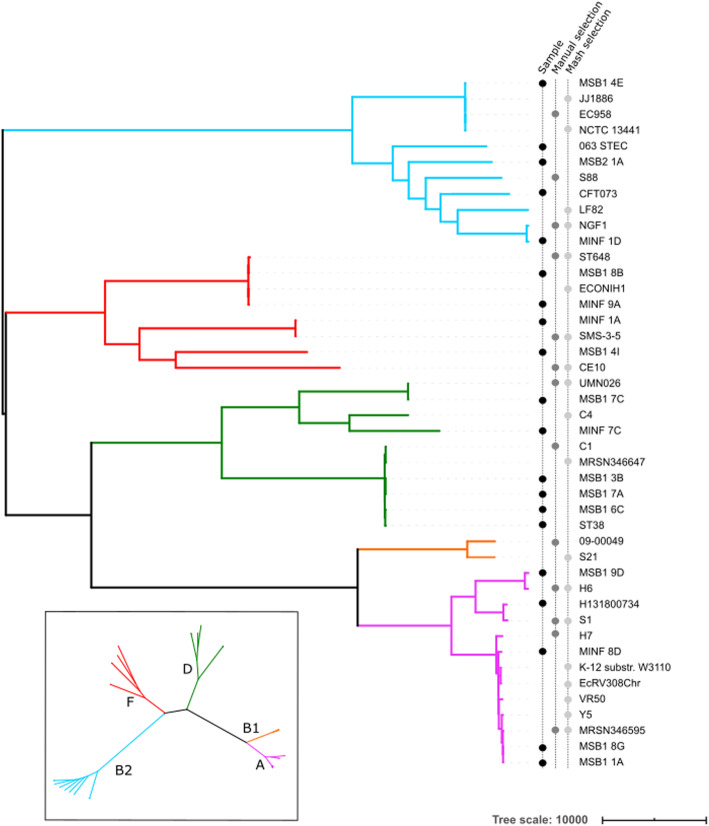
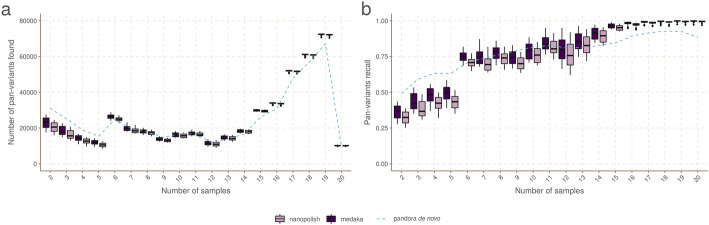
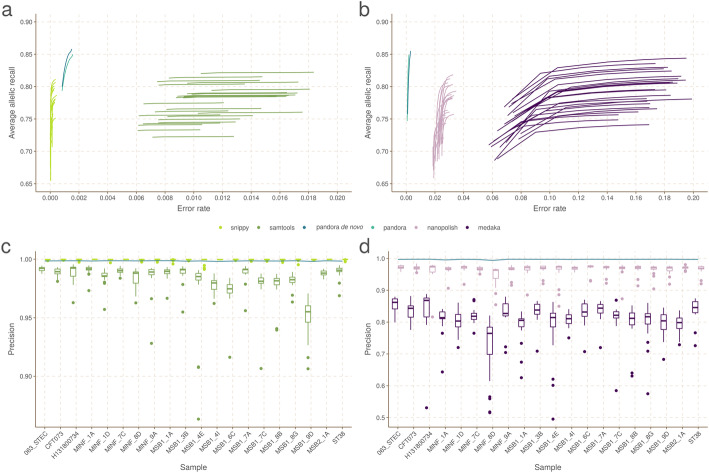
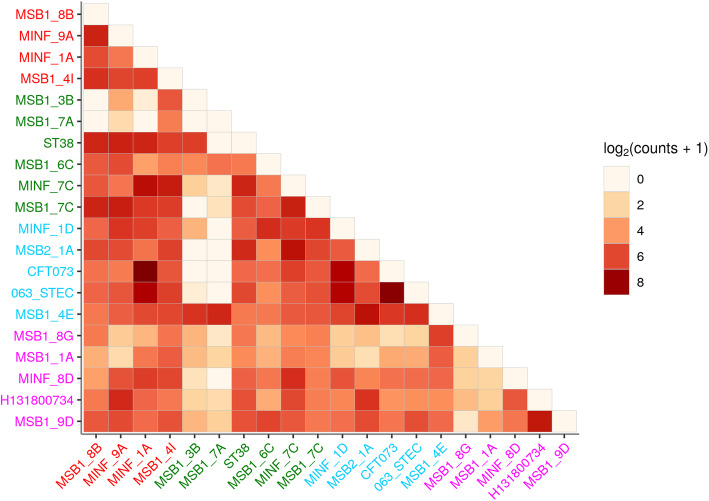
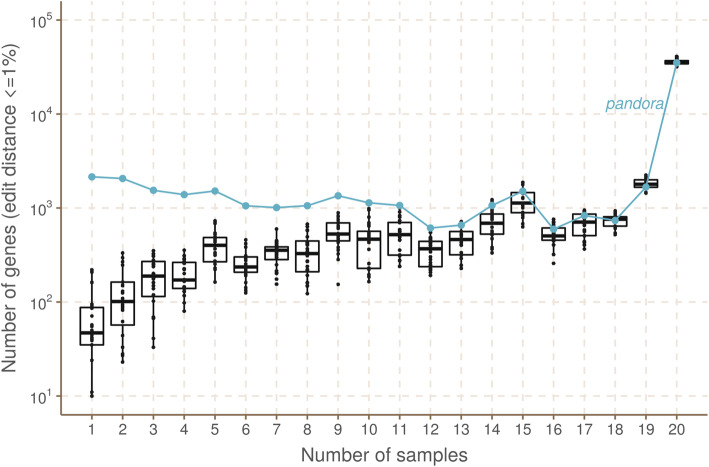
We present pandora, a novel pan-genome graph structure and algorithms for identifying variants across the full bacterial pan-genome. As much bacterial adaptability hinges on the accessory genome, methods which analyze SNPs in just the core genome have unsatisfactory limitations. Pandora approximates a sequenced genome as a recombinant of references, detects novel variation and pan-genotypes multiple samples. Using a reference graph of 578 Escherichia coli genomes, we compare 20 diverse isolates. Pandora recovers more rare SNPs than single-reference-based tools, is significantly better than picking the closest RefSeq reference, and provides a stable framework for analyzing diverse samples without reference bias.
Keywords: Accessory genome; Genome graph; Nanopore; Pan-genome.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests
Figures

References
-
- Lynch M, Ackerman MS, Gout J-F, Long H, Sung W, Thomas WK, et al. Genetic drift, selection and the evolution of the mutation rate. Nat Rev Genet. Nature Publishing Group. 2016;17(11):704–14. 10.1038/nrg.2016.104. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
