

Establishing the phenotypic spectrum of ZTTK syndrome by analysis of 52 individuals with variants in SON
- PMID: 34521999
- PMCID: PMC8904542
- DOI: 10.1038/s41431-021-00960-4
Establishing the phenotypic spectrum of ZTTK syndrome by analysis of 52 individuals with variants in SON
Abstract
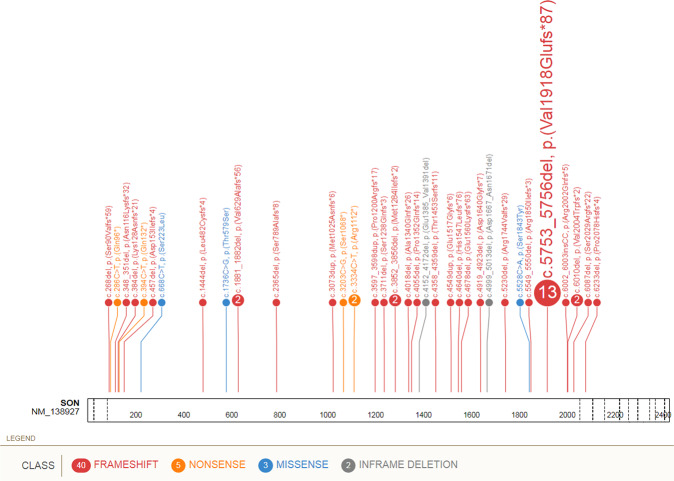
Zhu-Tokita-Takenouchi-Kim (ZTTK) syndrome, an intellectual disability syndrome first described in 2016, is caused by heterozygous loss-of-function variants in SON. Its encoded protein promotes pre-mRNA splicing of many genes essential for development. Whereas individual phenotypic traits have previously been linked to erroneous splicing of SON target genes, the phenotypic spectrum and the pathogenicity of missense variants have not been further evaluated. We present the phenotypic abnormalities in 52 individuals, including 17 individuals who have not been reported before. In total, loss-of-function variants were detected in 49 individuals (de novo in 47, inheritance unknown in 2), and in 3, a missense variant was observed (2 de novo, 1 inheritance unknown). Phenotypic abnormalities, systematically collected and analyzed in Human Phenotype Ontology, were found in all organ systems. Significant inter-individual phenotypic variability was observed, even in individuals with the same recurrent variant (n = 13). SON haploinsufficiency was previously shown to lead to downregulation of downstream genes, contributing to specific phenotypic features. Similar functional analysis for one missense variant, however, suggests a different mechanism than for heterozygous loss-of-function. Although small in numbers and while pathogenicity of these variants is not certain, these data allow for speculation whether de novo missense variants cause ZTTK syndrome via another mechanism, or a separate overlapping syndrome. In conclusion, heterozygous loss-of-function variants in SON define a recognizable syndrome, ZTTK, associated with a broad, severe phenotypic spectrum, characterized by a large inter-individual variability. These observations provide essential information for affected individuals, parents, and healthcare professionals to ensure appropriate clinical management.
© 2021. The Author(s), under exclusive licence to European Society of Human Genetics.
Conflict of interest statement
The authors declare no competing interests.
Figures


Comment in
-
Commentary on: Establishing the phenotypic spectrum of ZTTK syndrome by analysis of 52 individuals with variants in SON.Eur J Hum Genet. 2022 Mar;30(3):258-259. doi: 10.1038/s41431-021-01007-4. Epub 2021 Nov 29. Eur J Hum Genet. 2022. PMID: 34840333 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
