

Minimizer-space de Bruijn graphs: Whole-genome assembly of long reads in minutes on a personal computer
- PMID: 34525345
- PMCID: PMC8562525
- DOI: 10.1016/j.cels.2021.08.009
Minimizer-space de Bruijn graphs: Whole-genome assembly of long reads in minutes on a personal computer
Abstract
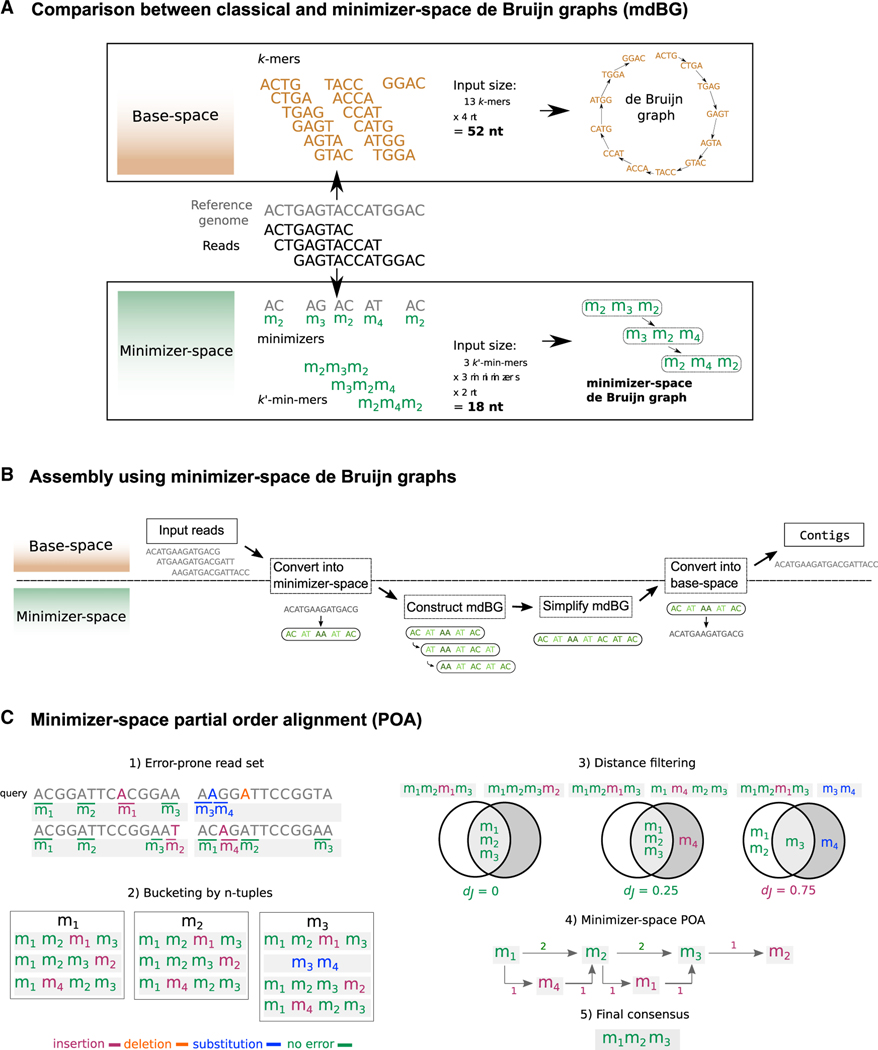
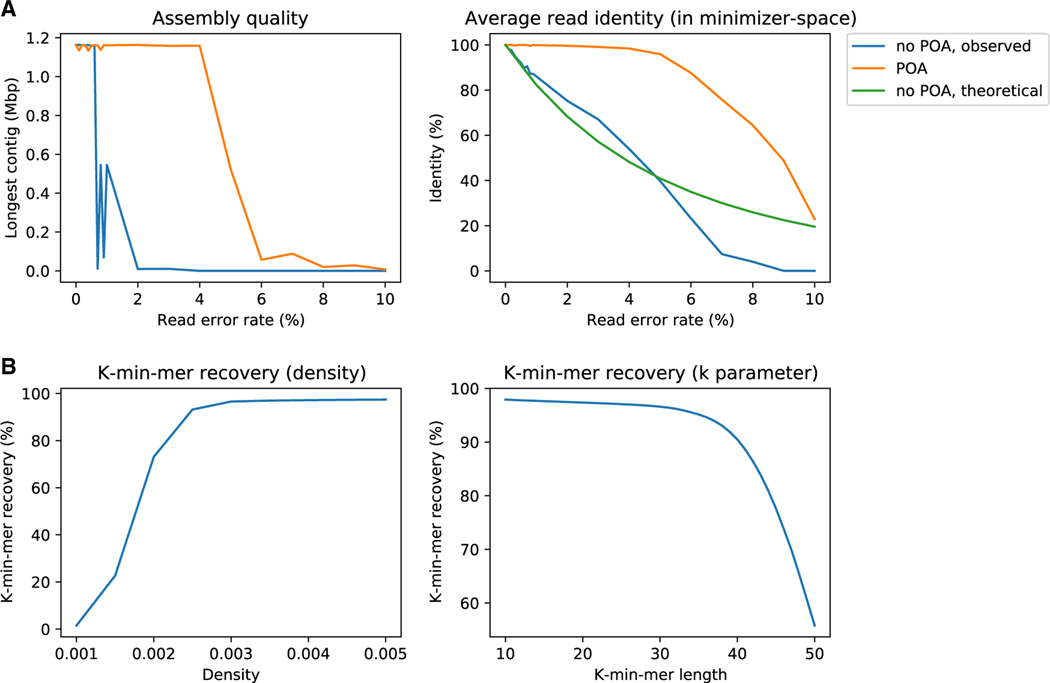
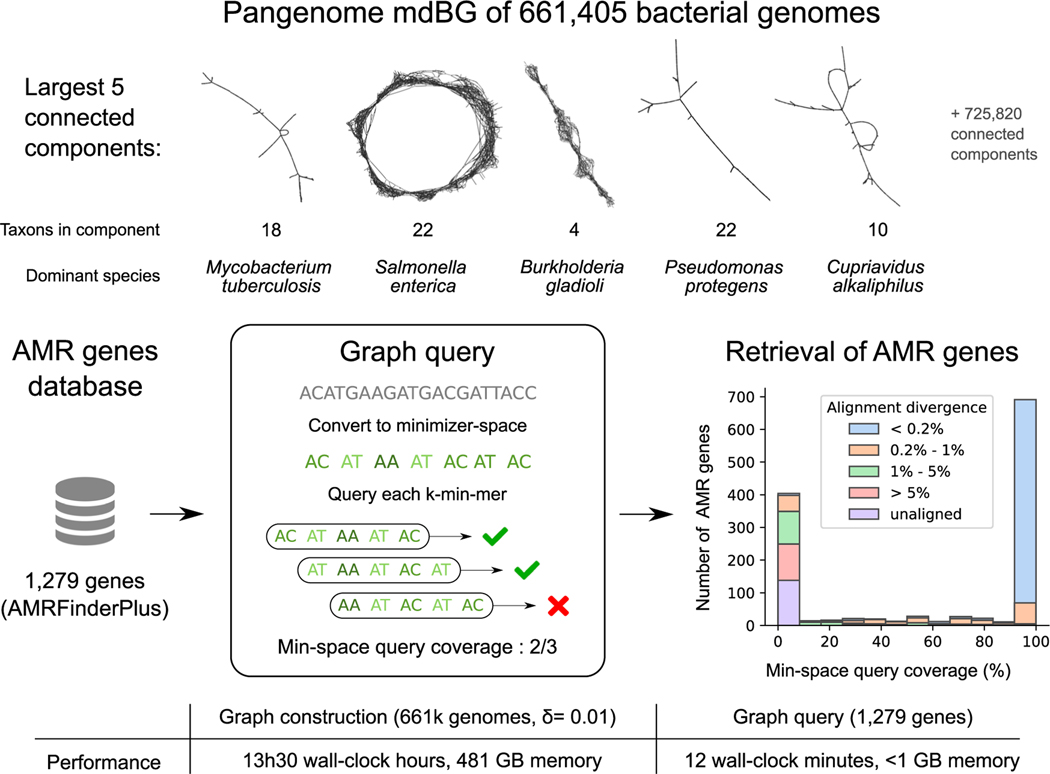
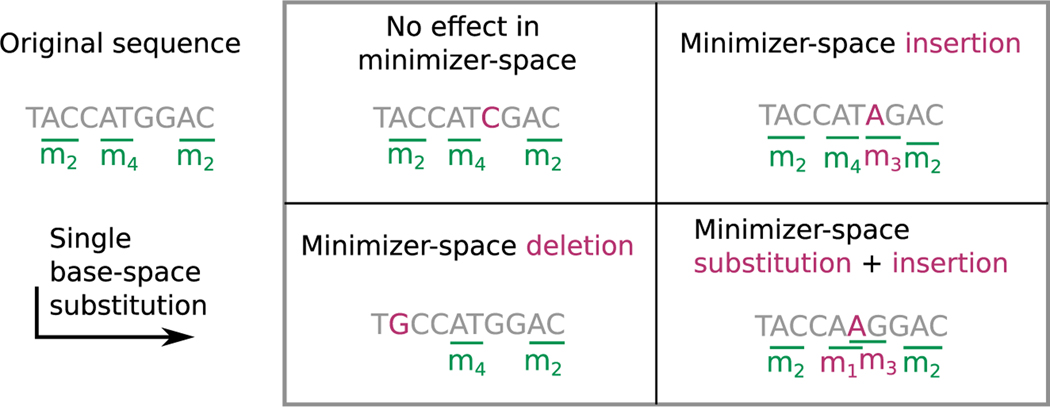
DNA sequencing data continue to progress toward longer reads with increasingly lower sequencing error rates. Here, we define an algorithmic approach, mdBG, that makes use of minimizer-space de Bruijn graphs to enable long-read genome assembly. mdBG achieves orders-of-magnitude improvement in both speed and memory usage over existing methods without compromising accuracy. A human genome is assembled in under 10 min using 8 cores and 10 GB RAM, and 60 Gbp of metagenome reads are assembled in 4 min using 1 GB RAM. In addition, we constructed a minimizer-space de Bruijn graph-based representation of 661,405 bacterial genomes, comprising 16 million nodes and 45 million edges, and successfully search it for anti-microbial resistance (AMR) genes in 12 min. We expect our advances to be essential to sequence analysis, given the rise of long-read sequencing in genomics, metagenomics, and pangenomics. Code for constructing mdBGs is freely available for download at https://github.com/ekimb/rust-mdbg/.
Keywords: bacterial genomes; data structures; de Bruijn graphs; genome assembly; genome graphs; long-read sequencing; metagenomics; minimizers; pangenomics; partial order alignment.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Comment in
-
Democratizing long-read genome assembly.Cell Syst. 2021 Oct 20;12(10):945-947. doi: 10.1016/j.cels.2021.09.010. Cell Syst. 2021. PMID: 34672955
References
-
- Batu T, Ergun F, and Şahinalp C. (2006). Oblivious string embeddings and edit distance approximations. In Proceedings of the Seventeenth Annual ACM-SIAM Symposium on Discrete Algorithms’, SODA ‘06, Society for Industrial and Applied Mathematics), pp. 792–801.
-
- Bingmann T, Bradley P, Gauger F, and Iqbal Z. (2019). COBS: a compact bit-sliced signature index. In 26th International Conference on String Processing and Information Retrieval (SPIRE), pp. 285–303. arXiv:1905. 09624v2.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
