

Translating genetic and functional data into clinical practice: a series of 223 families with myotonia
- PMID: 34529042
- PMCID: PMC9014745
- DOI: 10.1093/brain/awab344
Translating genetic and functional data into clinical practice: a series of 223 families with myotonia
Abstract
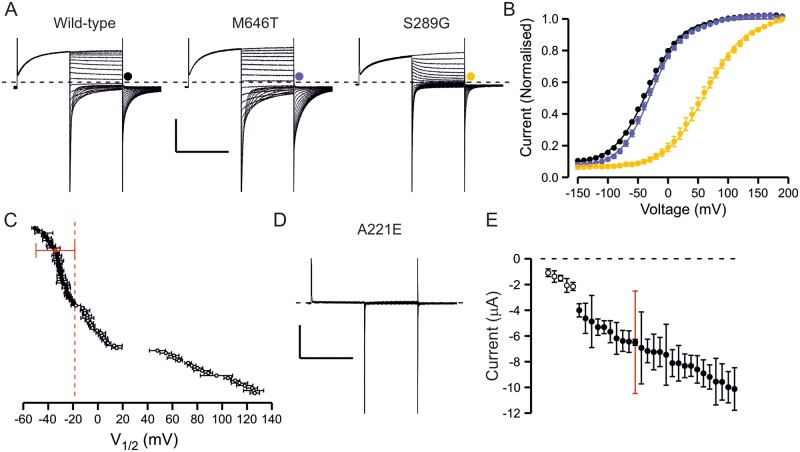
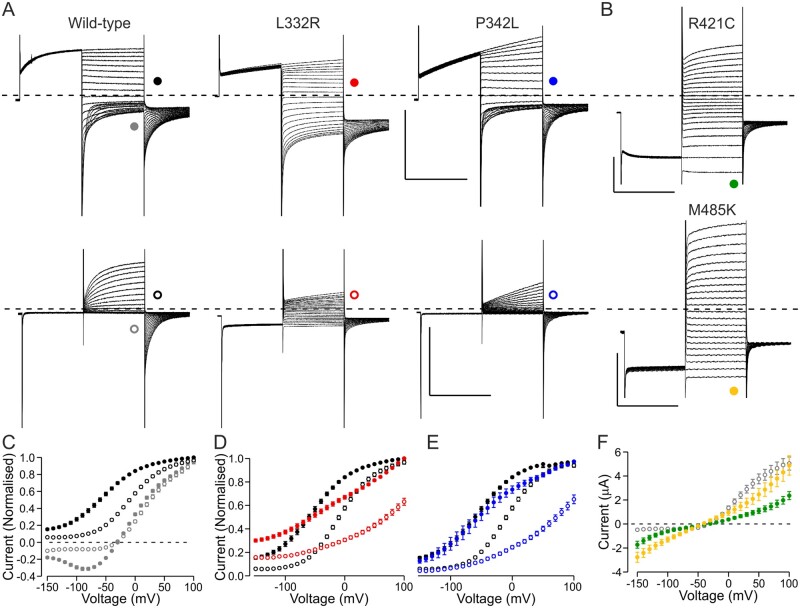
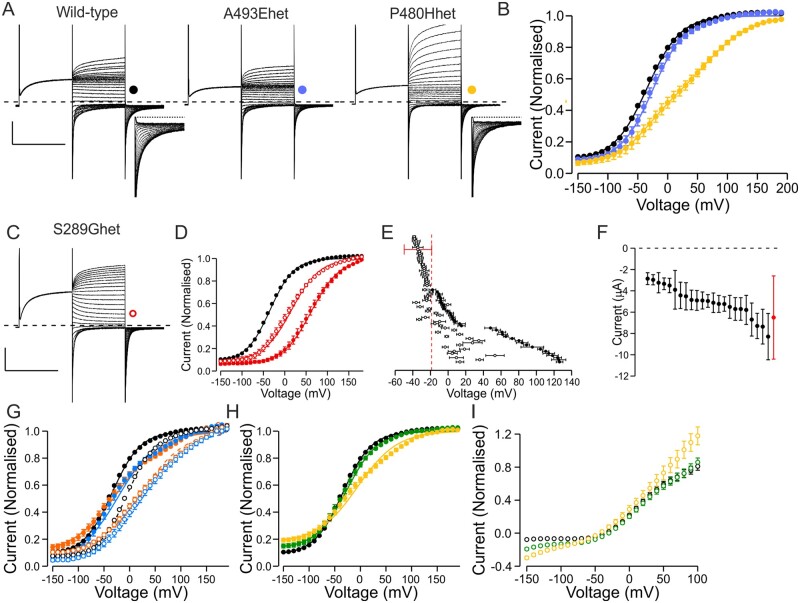
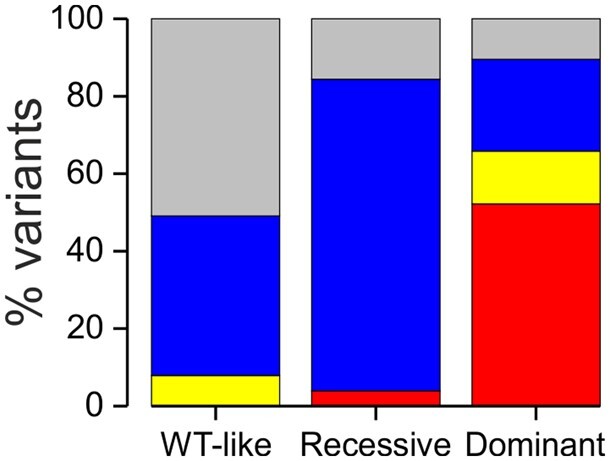
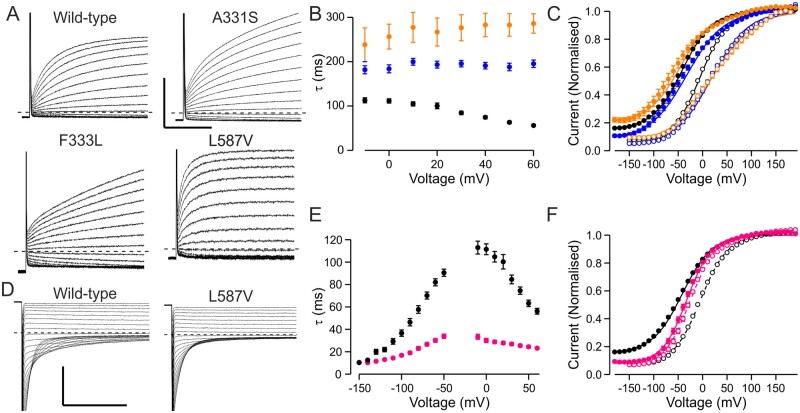
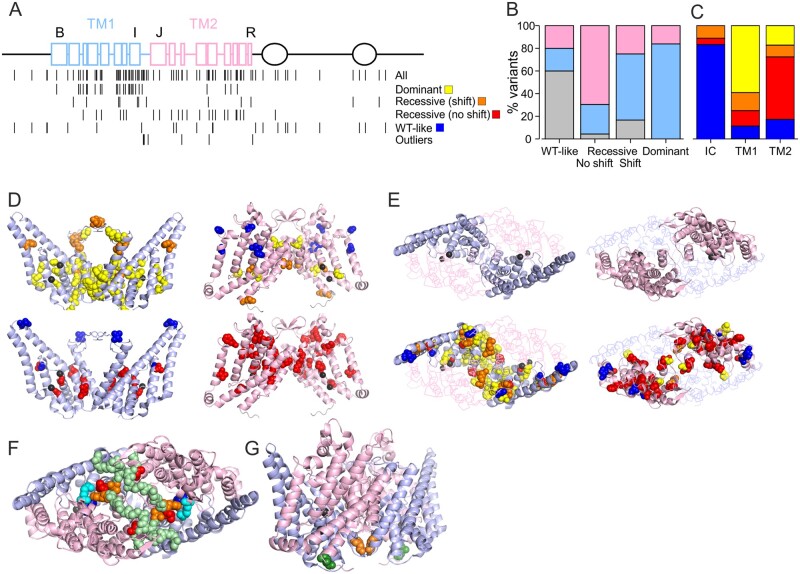
High-throughput DNA sequencing is increasingly employed to diagnose single gene neurological and neuromuscular disorders. Large volumes of data present new challenges in data interpretation and its useful translation into clinical and genetic counselling for families. Even when a plausible gene is identified with confidence, interpretation of the clinical significance and inheritance pattern of variants can be challenging. We report our approach to evaluating variants in the skeletal muscle chloride channel ClC-1 identified in 223 probands with myotonia congenita as an example of these challenges. Sequencing of CLCN1, the gene that encodes CLC-1, is central to the diagnosis of myotonia congenita. However, interpreting the pathogenicity and inheritance pattern of novel variants is notoriously difficult as both dominant and recessive mutations are reported throughout the channel sequence, ClC-1 structure-function is poorly understood and significant intra- and interfamilial variability in phenotype is reported. Heterologous expression systems to study functional consequences of CIC-1 variants are widely reported to aid the assessment of pathogenicity and inheritance pattern. However, heterogeneity of reported analyses does not allow for the systematic correlation of available functional and genetic data. We report the systematic evaluation of 95 CIC-1 variants in 223 probands, the largest reported patient cohort, in which we apply standardized functional analyses and correlate this with clinical assessment and inheritance pattern. Such correlation is important to determine whether functional data improves the accuracy of variant interpretation and likely mode of inheritance. Our data provide an evidence-based approach that functional characterization of ClC-1 variants improves clinical interpretation of their pathogenicity and inheritance pattern, and serve as reference for 34 previously unreported and 28 previously uncharacterized CLCN1 variants. In addition, we identify novel pathogenic mechanisms and find that variants that alter voltage dependence of activation cluster in the first half of the transmembrane domains and variants that yield no currents cluster in the second half of the transmembrane domain. None of the variants in the intracellular domains were associated with dominant functional features or dominant inheritance pattern of myotonia congenita. Our data help provide an initial estimate of the anticipated inheritance pattern based on the location of a novel variant and shows that systematic functional characterization can significantly refine the assessment of risk of an associated inheritance pattern and consequently the clinical and genetic counselling.
Keywords: CLCN1; ClC-1; chloride channel; myotonia congenita; skeletal muscle channelopathy.
© The Author(s) (2021). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
Similar articles
-
Chloride channel myotonia: exon 8 hot-spot for dominant-negative interactions.Brain. 2007 Dec;130(Pt 12):3265-74. doi: 10.1093/brain/awm248. Epub 2007 Oct 11. Brain. 2007. PMID: 17932099
-
ClC-1 mutations in myotonia congenita patients: insights into molecular gating mechanisms and genotype-phenotype correlation.J Physiol. 2015 Sep 15;593(18):4181-99. doi: 10.1113/JP270358. Epub 2015 Jul 14. J Physiol. 2015. PMID: 26096614 Free PMC article.
-
Clinical, Molecular, and Functional Characterization of CLCN1 Mutations in Three Families with Recessive Myotonia Congenita.Neuromolecular Med. 2015 Sep;17(3):285-96. doi: 10.1007/s12017-015-8356-8. Epub 2015 May 26. Neuromolecular Med. 2015. PMID: 26007199 Free PMC article.
-
Defective Gating and Proteostasis of Human ClC-1 Chloride Channel: Molecular Pathophysiology of Myotonia Congenita.Front Neurol. 2020 Feb 11;11:76. doi: 10.3389/fneur.2020.00076. eCollection 2020. Front Neurol. 2020. PMID: 32117034 Free PMC article. Review.
-
Physiology and pathophysiology of CLC-1: mechanisms of a chloride channel disease, myotonia.J Biomed Biotechnol. 2011;2011:685328. doi: 10.1155/2011/685328. Epub 2011 Dec 1. J Biomed Biotechnol. 2011. PMID: 22187529 Free PMC article. Review.
Cited by
-
Deciphering the Genetic Basis of Degenerative and Developmental Eye Disorders in 50 Pakistani Consanguineous Families Using Whole-Exome Sequencing.Int J Mol Sci. 2025 Mar 18;26(6):2715. doi: 10.3390/ijms26062715. Int J Mol Sci. 2025. PMID: 40141357 Free PMC article.
-
Clinical and Genetic Spectrum of Myotonia Congenita in Turkish Children.J Neuromuscul Dis. 2023;10(5):915-924. doi: 10.3233/JND-230046. J Neuromuscul Dis. 2023. PMID: 37355912 Free PMC article.
-
Periodic paralysis.Handb Clin Neurol. 2024;203:39-58. doi: 10.1016/B978-0-323-90820-7.00002-1. Handb Clin Neurol. 2024. PMID: 39174253 Free PMC article. Review.
-
Maternal obesity and ancestry distance in influencing birth outcomes.Int J Obes (Lond). 2025 Apr 12. doi: 10.1038/s41366-025-01783-9. Online ahead of print. Int J Obes (Lond). 2025. PMID: 40221546
-
ClC-1 Chloride Channel: Inputs on the Structure-Function Relationship of Myotonia Congenita-Causing Mutations.Biomedicines. 2023 Sep 24;11(10):2622. doi: 10.3390/biomedicines11102622. Biomedicines. 2023. PMID: 37892996 Free PMC article. Review.
References
-
- Koch MC, Steinmeyer K, Lorenz C, et al. . The skeletal muscle chloride channel in dominant and recessive human myotonia. Science (New York, NY). 1992;257(5071):797–800. - PubMed
-
- George AL Jr, Crackower MA, Abdalla JA, Hudson AJ, Ebers GC.. Molecular basis of Thomsen's disease (autosomal dominant myotonia congenita). Nat Genet. 1993;3(4):305–310. - PubMed
-
- Bugiardini E, Rivolta I, Binda A, et al. . SCN4A mutation as modifying factor of myotonic dystrophy type 2 phenotype. Neuromuscul Disord. 2015;25(4):301–307. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
