

Identification of tissue-specific and common methylation quantitative trait loci in healthy individuals using MAGAR
- PMID: 34530905
- PMCID: PMC8444396
- DOI: 10.1186/s13072-021-00415-6
Identification of tissue-specific and common methylation quantitative trait loci in healthy individuals using MAGAR
Abstract
Background: Understanding the influence of genetic variants on DNA methylation is fundamental for the interpretation of epigenomic data in the context of disease. There is a need for systematic approaches not only for determining methylation quantitative trait loci (methQTL), but also for discriminating general from cell type-specific effects.
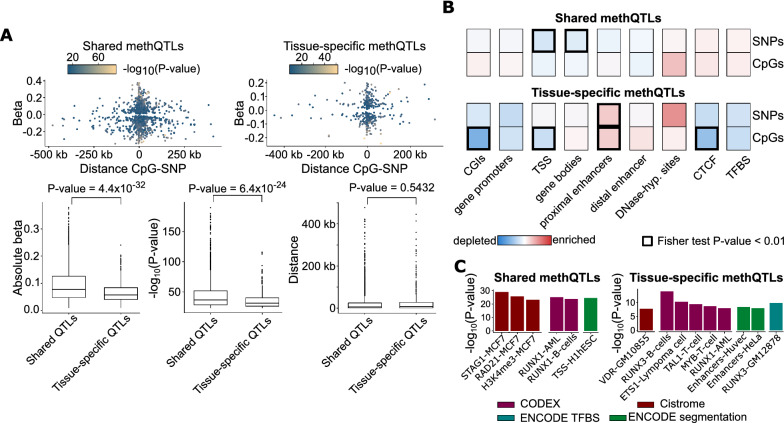
Results: Here, we present a two-step computational framework MAGAR ( https://bioconductor.org/packages/MAGAR ), which fully supports the identification of methQTLs from matched genotyping and DNA methylation data, and additionally allows for illuminating cell type-specific methQTL effects. In a pilot analysis, we apply MAGAR on data in four tissues (ileum, rectum, T cells, B cells) from healthy individuals and demonstrate the discrimination of common from cell type-specific methQTLs. We experimentally validate both types of methQTLs in an independent data set comprising additional cell types and tissues. Finally, we validate selected methQTLs located in the PON1, ZNF155, and NRG2 genes by ultra-deep local sequencing. In line with previous reports, we find cell type-specific methQTLs to be preferentially located in enhancer elements.
Conclusions: Our analysis demonstrates that a systematic analysis of methQTLs provides important new insights on the influences of genetic variants to cell type-specific epigenomic variation.
Keywords: Computational biology; DNA methylation; Quantitative trait loci; Tissue specificity.
© 2021. The Author(s).
Conflict of interest statement
Y.S.A. is a co-founder and co-owner of PolyKnomics, a private organization active in the fields of quantitative, computational, and statistical genomics. All other authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
