

Proteogenomic characterization of pancreatic ductal adenocarcinoma
- PMID: 34534465
- PMCID: PMC8654574
- DOI: 10.1016/j.cell.2021.08.023
Proteogenomic characterization of pancreatic ductal adenocarcinoma
Abstract
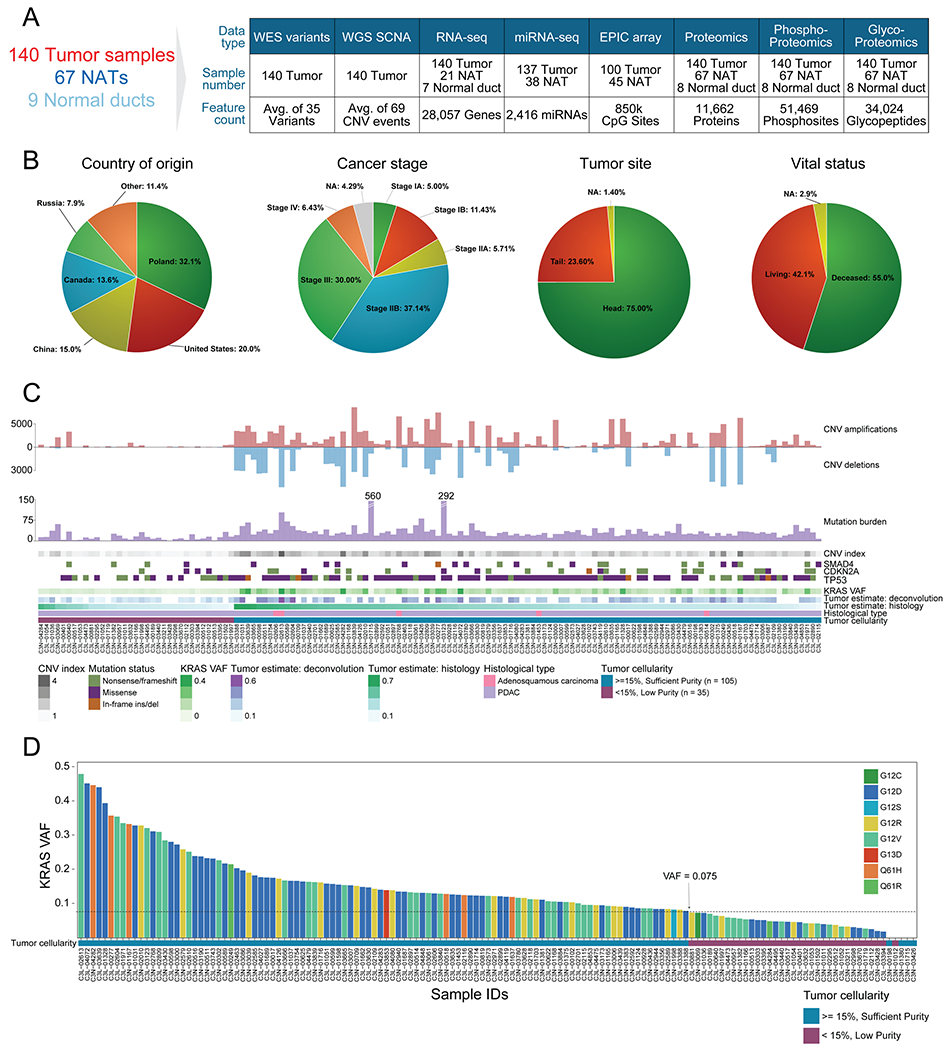
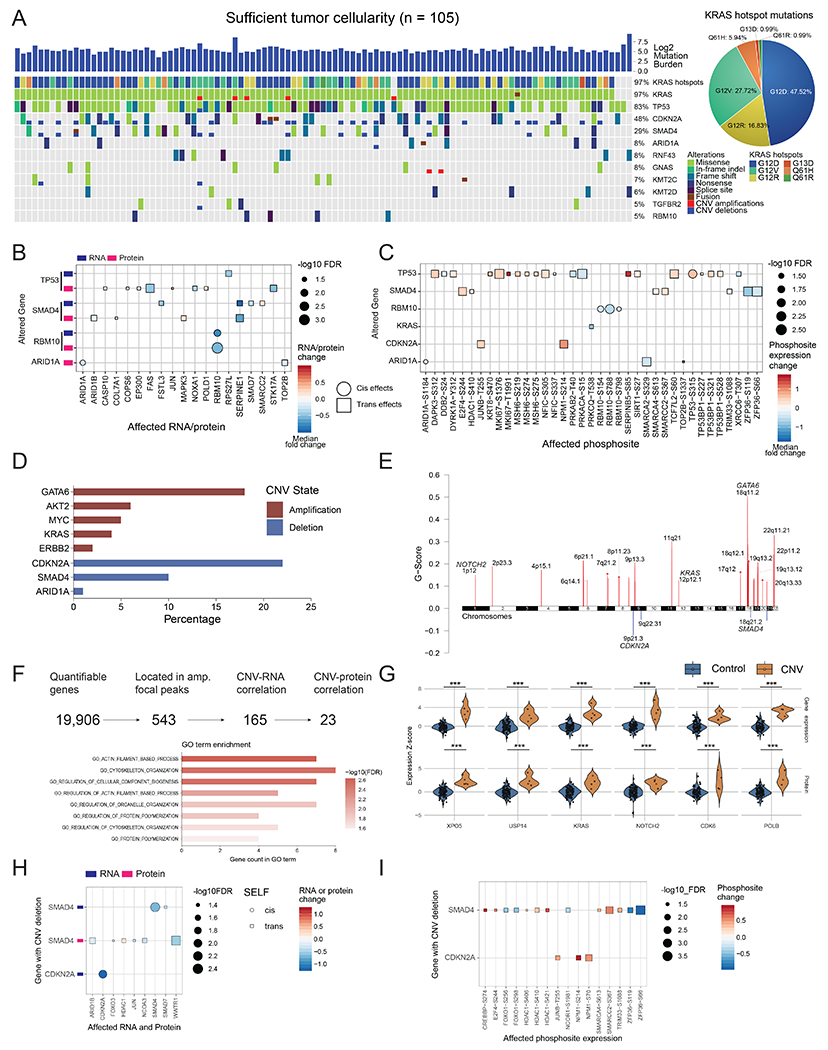
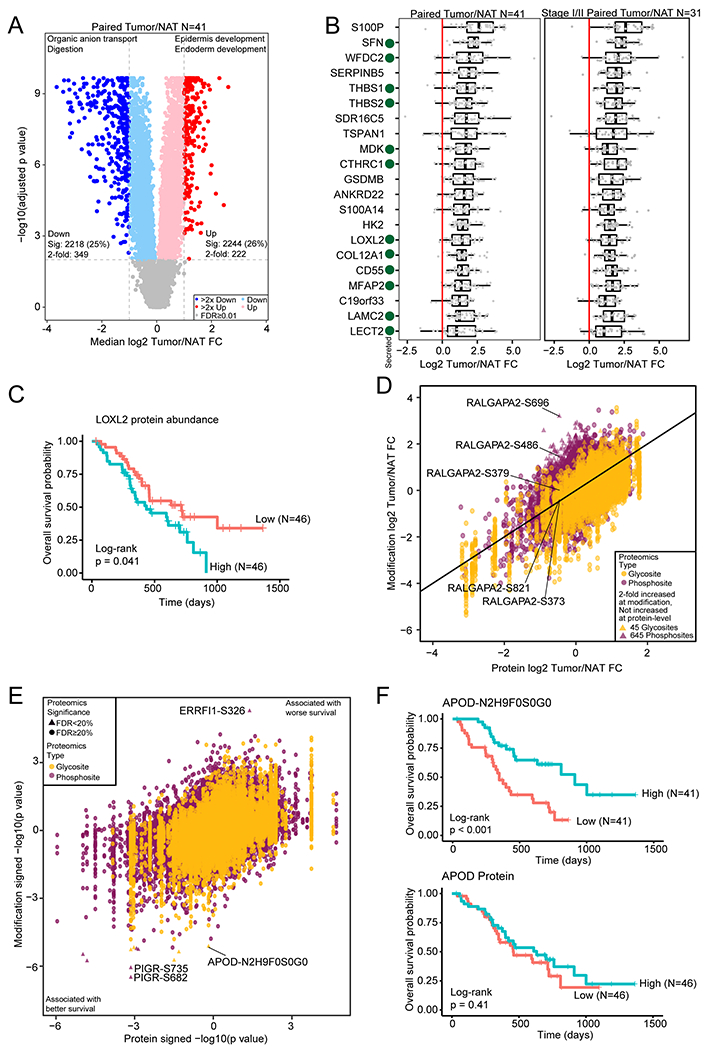
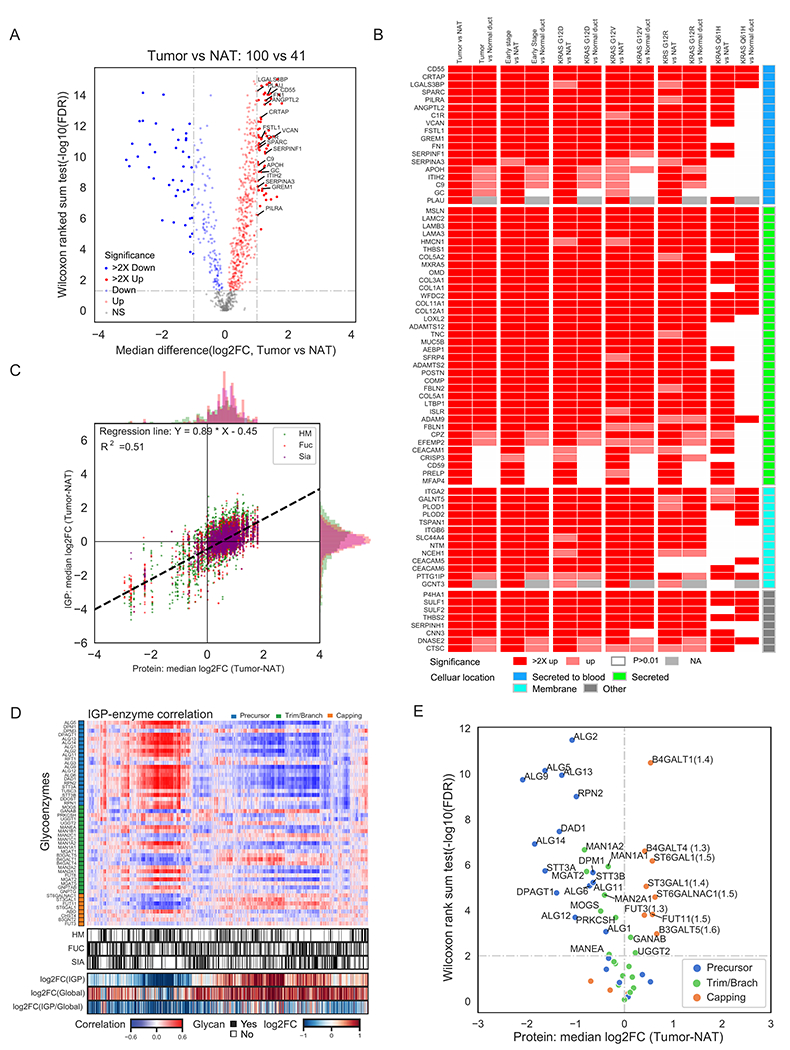
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive cancer with poor patient survival. Toward understanding the underlying molecular alterations that drive PDAC oncogenesis, we conducted comprehensive proteogenomic analysis of 140 pancreatic cancers, 67 normal adjacent tissues, and 9 normal pancreatic ductal tissues. Proteomic, phosphoproteomic, and glycoproteomic analyses were used to characterize proteins and their modifications. In addition, whole-genome sequencing, whole-exome sequencing, methylation, RNA sequencing (RNA-seq), and microRNA sequencing (miRNA-seq) were performed on the same tissues to facilitate an integrated proteogenomic analysis and determine the impact of genomic alterations on protein expression, signaling pathways, and post-translational modifications. To ensure robust downstream analyses, tumor neoplastic cellularity was assessed via multiple orthogonal strategies using molecular features and verified via pathological estimation of tumor cellularity based on histological review. This integrated proteogenomic characterization of PDAC will serve as a valuable resource for the community, paving the way for early detection and identification of novel therapeutic targets.
Keywords: CPTAC; KRAS; endothelial cell; glycoproteins; immune-cold tumors; kinase inhibitors; neoplastic cellularity; pancreatic ductal adenocarcinoma; proteogenomics; tumor subtyping.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests R.H.H. has the potential of receiving royalty payments from Thrive Earlier Diagnosis for the GNAS invention in a relationship overseen by Johns Hopkins University. The remaining authors declare no competing interests.
Figures
References
-
- Akagi J, Takai E, Tamori Y, Nakagawa K, and Ogawa M (2001). CA19-9 epitope a possible marker for MUC-1/Y protein. International journal of oncology 18, 1085–1091. - PubMed
-
- American Cancer Society (2021). Cancer Facts & Figures 2021. Atlanta: American Cancer Society.
SI Reference
-
- Arntfield ME, and van der Kooy D (2011). beta-Cell evolution: How the pancreas borrowed from the brain: The shared toolbox of genes expressed by neural and pancreatic endocrine cells may reflect their evolutionary relationship. Bioessays 33, 582–587. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U24 CA210954/CA/NCI NIH HHS/United States
- P30 ES010126/ES/NIEHS NIH HHS/United States
- U01 CA214116/CA/NCI NIH HHS/United States
- U24 CA210985/CA/NCI NIH HHS/United States
- U24 CA210986/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- U24 CA210972/CA/NCI NIH HHS/United States
- U24 CA210955/CA/NCI NIH HHS/United States
- P30 ES017885/ES/NIEHS NIH HHS/United States
- U24 CA210993/CA/NCI NIH HHS/United States
- U24 CA210979/CA/NCI NIH HHS/United States
- R50 CA243627/CA/NCI NIH HHS/United States
- U24 CA210967/CA/NCI NIH HHS/United States
- U01 CA214114/CA/NCI NIH HHS/United States
- U01 CA214125/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
