

MAPK-pathway inhibition mediates inflammatory reprogramming and sensitizes tumors to targeted activation of innate immunity sensor RIG-I
- PMID: 34535668
- PMCID: PMC8448826
- DOI: 10.1038/s41467-021-25728-8
MAPK-pathway inhibition mediates inflammatory reprogramming and sensitizes tumors to targeted activation of innate immunity sensor RIG-I
Abstract
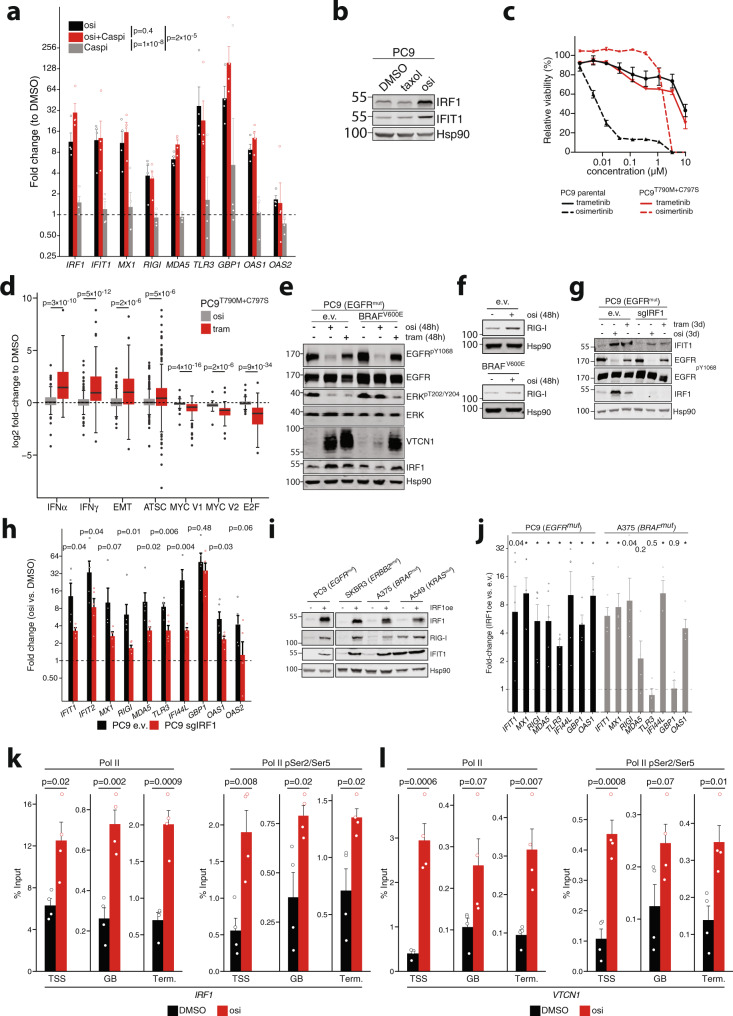
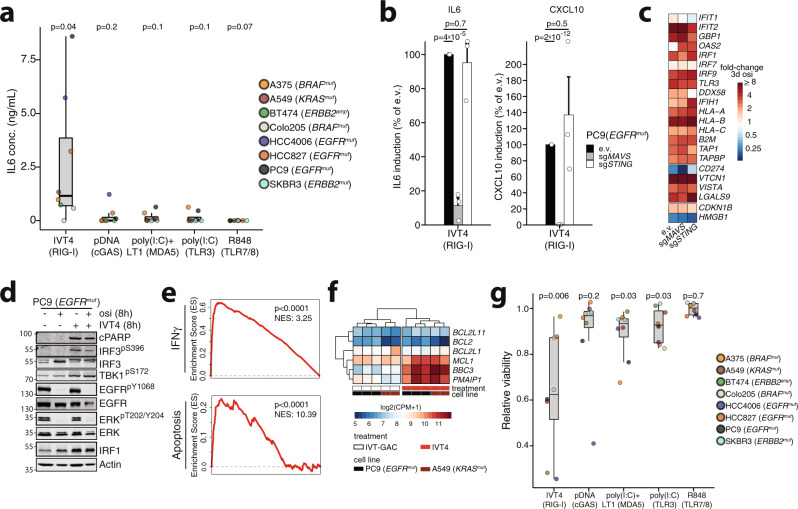
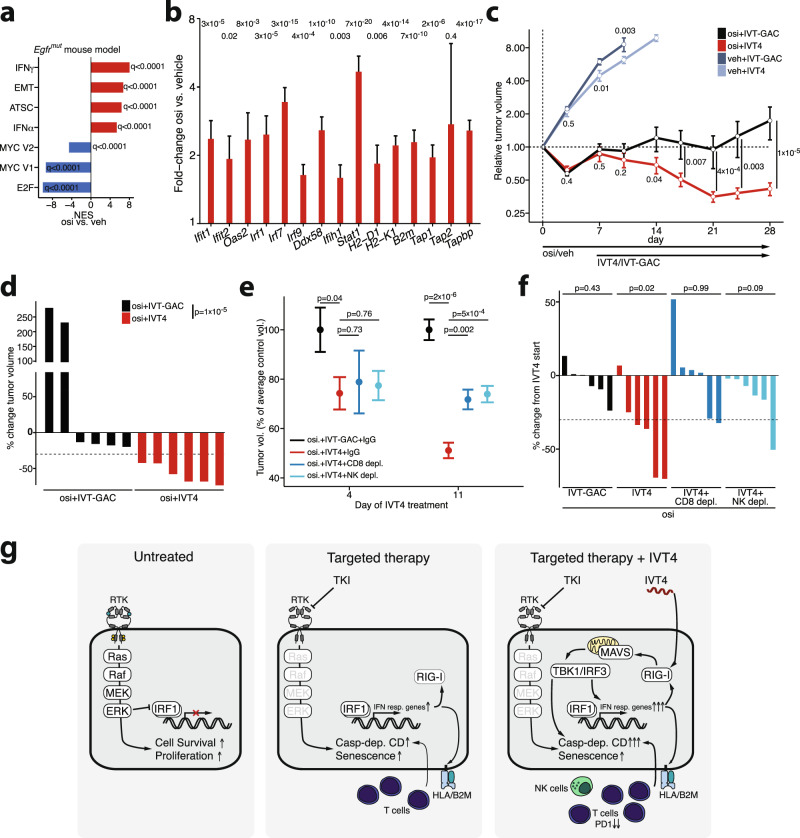
Kinase inhibitors suppress the growth of oncogene driven cancer but also enforce the selection of treatment resistant cells that are thought to promote tumor relapse in patients. Here, we report transcriptomic and functional genomics analyses of cells and tumors within their microenvironment across different genotypes that persist during kinase inhibitor treatment. We uncover a conserved, MAPK/IRF1-mediated inflammatory response in tumors that undergo stemness- and senescence-associated reprogramming. In these tumor cells, activation of the innate immunity sensor RIG-I via its agonist IVT4, triggers an interferon and a pro-apoptotic response that synergize with concomitant kinase inhibition. In humanized lung cancer xenografts and a syngeneic Egfr-driven lung cancer model these effects translate into reduction of exhausted CD8+ T cells and robust tumor shrinkage. Overall, the mechanistic understanding of MAPK/IRF1-mediated intratumoral reprogramming may ultimately prolong the efficacy of targeted drugs in genetically defined cancer patients.
© 2021. The Author(s).
Conflict of interest statement
M.L.S. is a founder and shareholder of PearlRiver Bio (now part of Centessa Pharmaceuticals) and received consulting honoraria from PearlRiver Bio. M.L.S. receives research funding from PearlRiver Bio and Novartis. R.B. is an employee of Targos Molecular Pathology. H.L. is an employee of CrownBiosciences. G.H. is co‐founder of Rigontec GmbH. M.S. is listed as inventor on a patent application covering RIG-I activating structures. R.K.T. is founder of PearlRiver Bio (now part of Centessa Pharmaceuticals), founder of NEO New Oncology (now part of Siemens Healthcare), consulting honoraria from PearlRiver Bio and NEO New Oncology. K.O. received research funding from Boehringer Ingelheim, Novartis, AstraZeneca, Eli Lilly, and Daiichi-Sankyo outside the submitted work. K.O. reports honoraria from AstraZeneca, MSD and Chugai pharmaceutical outside the submitted work. The other authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
