

Role of genetics in amyotrophic lateral sclerosis: a large cohort study in Chinese mainland population
- PMID: 34544842
- PMCID: PMC9411893
- DOI: 10.1136/jmedgenet-2021-107965
Role of genetics in amyotrophic lateral sclerosis: a large cohort study in Chinese mainland population
Abstract
Background: A large number of new causative and risk genes for amyotrophic lateral sclerosis (ALS) have been identified mostly in patients of European ancestry. In contrast, we know relatively little regarding the genetics of ALS in other ethnic populations. This study aims to provide a comprehensive analysis of the genetics of ALS in an unprecedented large cohort of Chinese mainland population and correlate with the clinical features of rare variants carriers.
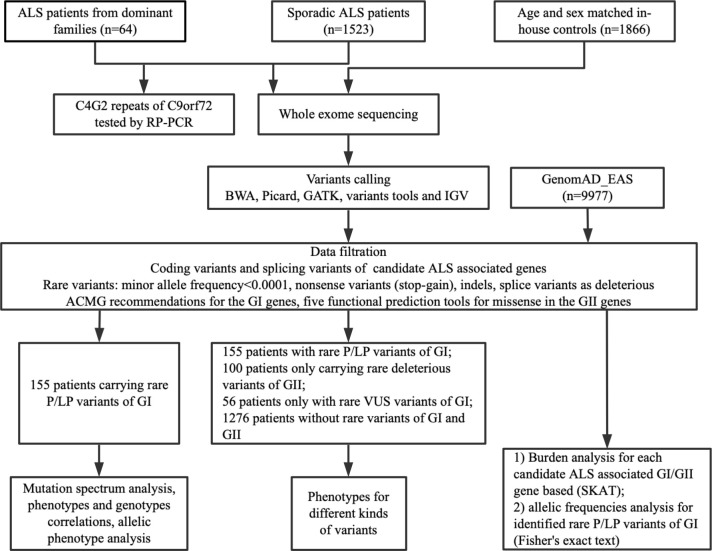
Methods: A total of 1587 patients, including 64 familial ALS (FALS) and 1523 sporadic ALS (SALS), and 1866 in-house controls were analysed by whole-exome sequencing and/or testing for G4C2 repeats in C9orf72. Forty-one ALS-associated genes were analysed.
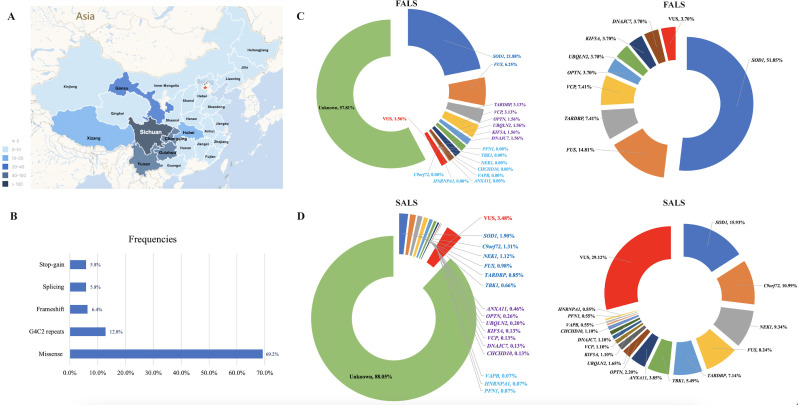
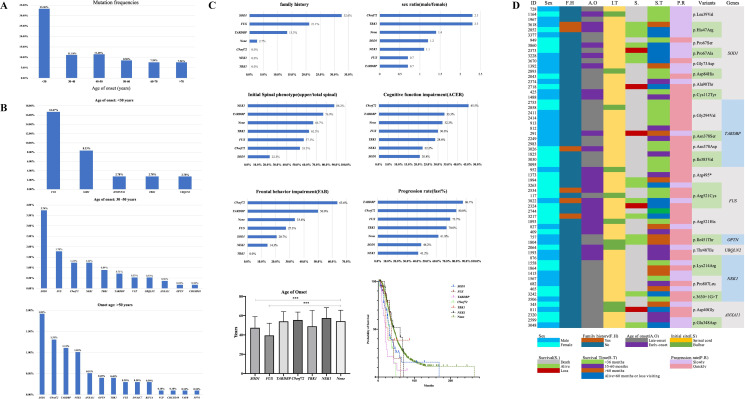
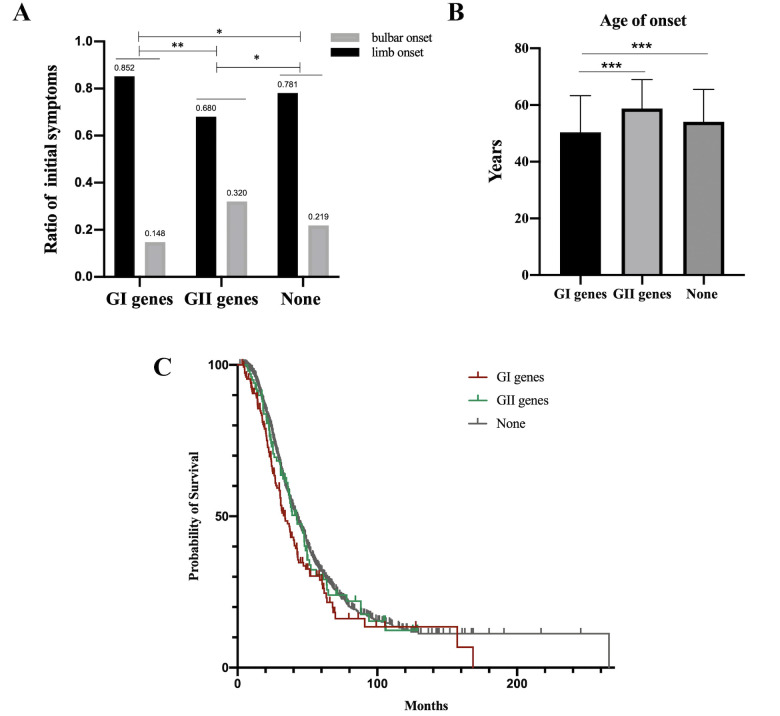
Findings: 155 patients, including 26 (40.6%) FALS and 129 (8.5%) SALS, carrying rare pathogenic/likely pathogenic (P/LP) variants of ALS causative genes were identified. SOD1 was the most common mutated gene, followed by C9orf72, FUS, NEK1, TARDBP and TBK1. By burden analysis, rare variants in SOD1, FUS and TARDBP contributed to the collective risk for ALS (p<2.5e-6) at the gene level, but at the allelic level TARDBP p.Gly294Val and FUS p.Arg521Cys and p.Arg521His were the most important single variants causing ALS. Clinically, P/LP variants in TARDBP and C9orf72 were associated with poor prognosis, in FUS linked with younger age of onset, and C9orf72 repeats tended to affect cognition.
Conclusions: Our data provide essential information for understanding the genetic and clinical features of ALS in China and for optimal design of genetic testing and evaluation of disease prognosis.
Keywords: genetic variation; genetics; medical; neurodegenerative diseases.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
References
-
- Brooks BR, Miller RG, Swash M, Munsat TL. World federation of neurology research group on motor neuron D. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–9. 10.1080/146608200300079536 - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous