

Rare and de novo variants in 827 congenital diaphragmatic hernia probands implicate LONP1 as candidate risk gene
- PMID: 34547244
- PMCID: PMC8546037
- DOI: 10.1016/j.ajhg.2021.08.011
Rare and de novo variants in 827 congenital diaphragmatic hernia probands implicate LONP1 as candidate risk gene
Abstract
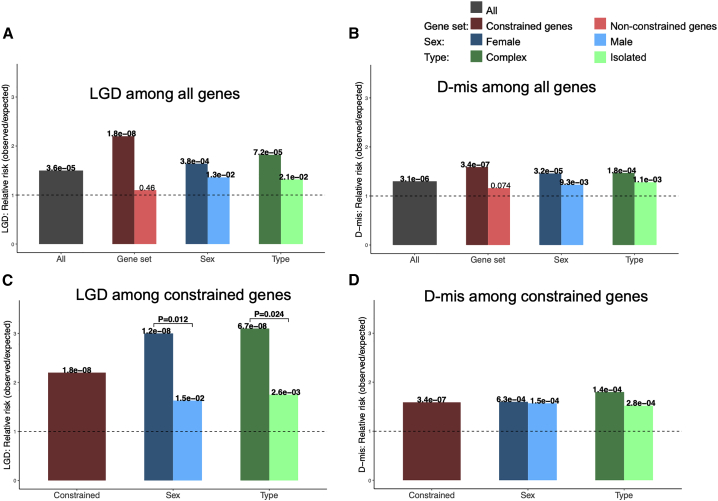
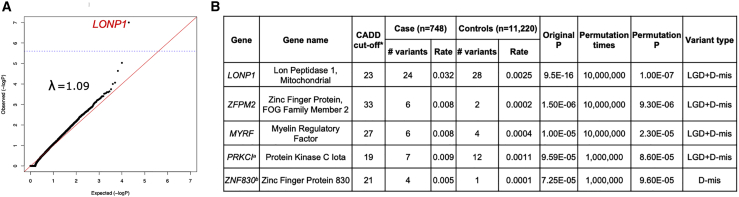
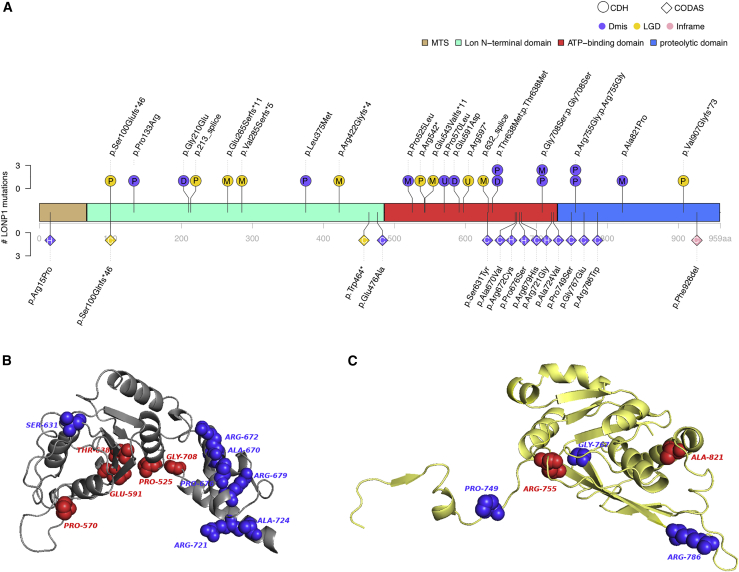
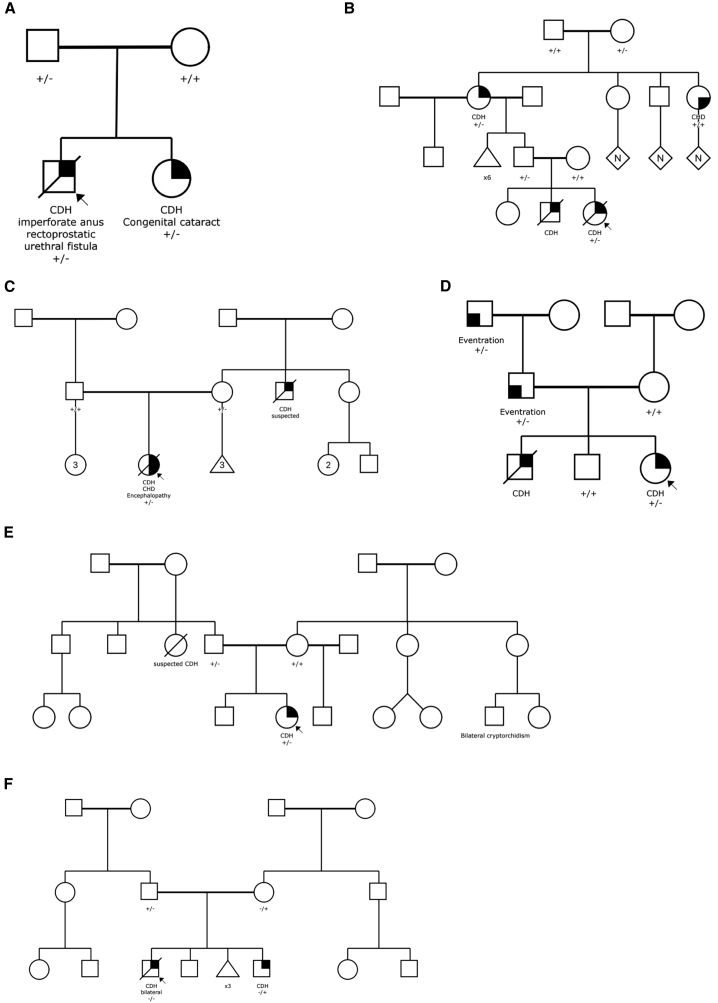
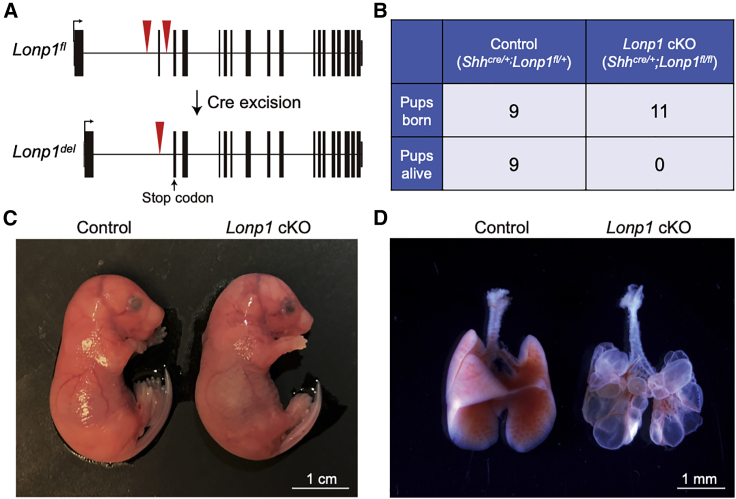
Congenital diaphragmatic hernia (CDH) is a severe congenital anomaly that is often accompanied by other anomalies. Although the role of genetics in the pathogenesis of CDH has been established, only a small number of disease-associated genes have been identified. To further investigate the genetics of CDH, we analyzed de novo coding variants in 827 proband-parent trios and confirmed an overall significant enrichment of damaging de novo variants, especially in constrained genes. We identified LONP1 (lon peptidase 1, mitochondrial) and ALYREF (Aly/REF export factor) as candidate CDH-associated genes on the basis of de novo variants at a false discovery rate below 0.05. We also performed ultra-rare variant association analyses in 748 affected individuals and 11,220 ancestry-matched population control individuals and identified LONP1 as a risk gene contributing to CDH through both de novo and ultra-rare inherited largely heterozygous variants clustered in the core of the domains and segregating with CDH in affected familial individuals. Approximately 3% of our CDH cohort who are heterozygous with ultra-rare predicted damaging variants in LONP1 have a range of clinical phenotypes, including other anomalies in some individuals and higher mortality and requirement for extracorporeal membrane oxygenation. Mice with lung epithelium-specific deletion of Lonp1 die immediately after birth, most likely because of the observed severe reduction of lung growth, a known contributor to the high mortality in humans. Our findings of both de novo and inherited rare variants in the same gene may have implications in the design and analysis for other genetic studies of congenital anomalies.
Keywords: ALYREF; LONP1; congenital diaphragmatic hernia; de novo variants.
Copyright © 2021 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Montalva L., Lauriti G., Zani A. Congenital heart disease associated with congenital diaphragmatic hernia: A systematic review on incidence, prenatal diagnosis, management, and outcome. J. Pediatr. Surg. 2019;54:909–919. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
