

Phylogeographic Clustering Suggests that Distinct Clades of Salmonella enterica Serovar Mississippi Are Endemic in Australia, the United Kingdom, and the United States
- PMID: 34550008
- PMCID: PMC8550085
- DOI: 10.1128/mSphere.00485-21
Phylogeographic Clustering Suggests that Distinct Clades of Salmonella enterica Serovar Mississippi Are Endemic in Australia, the United Kingdom, and the United States
Abstract
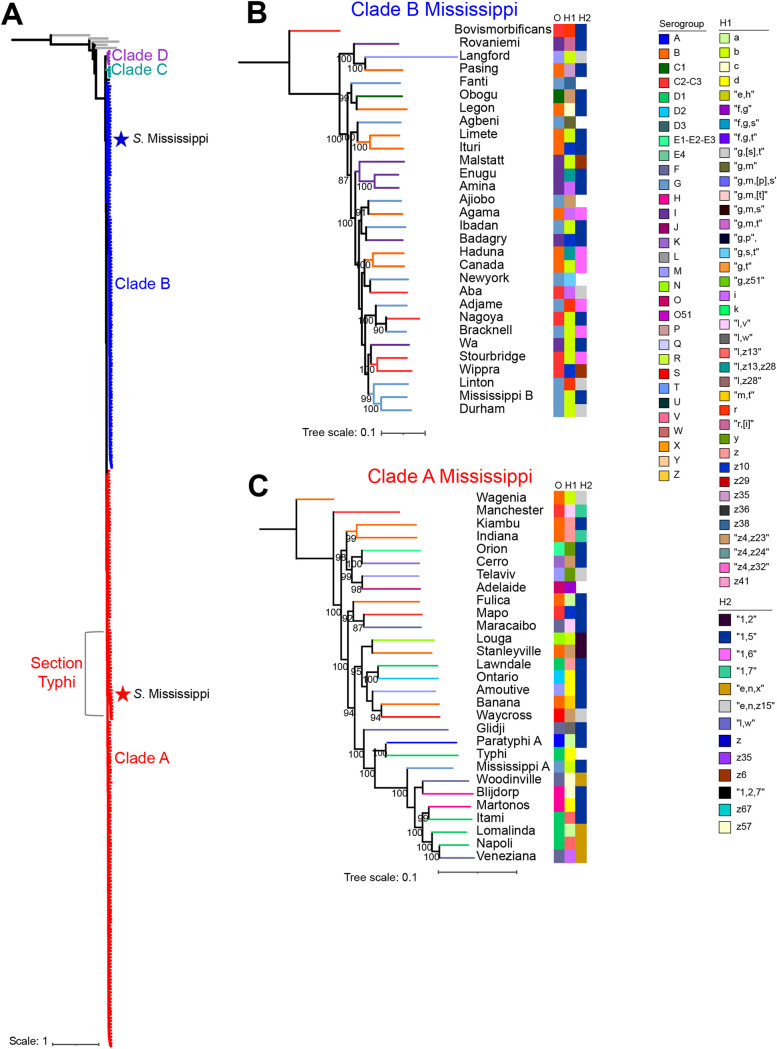
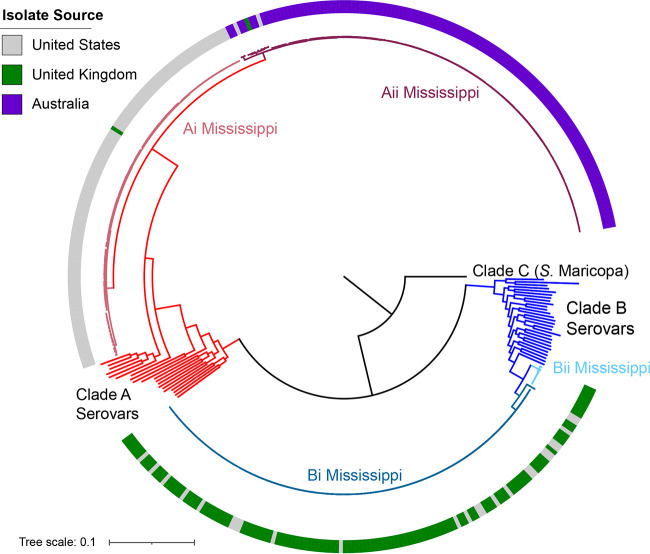
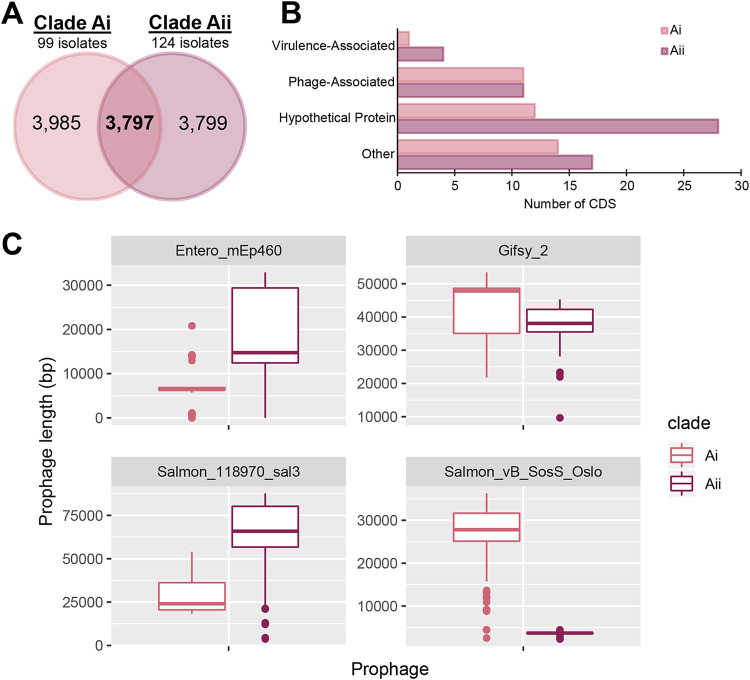
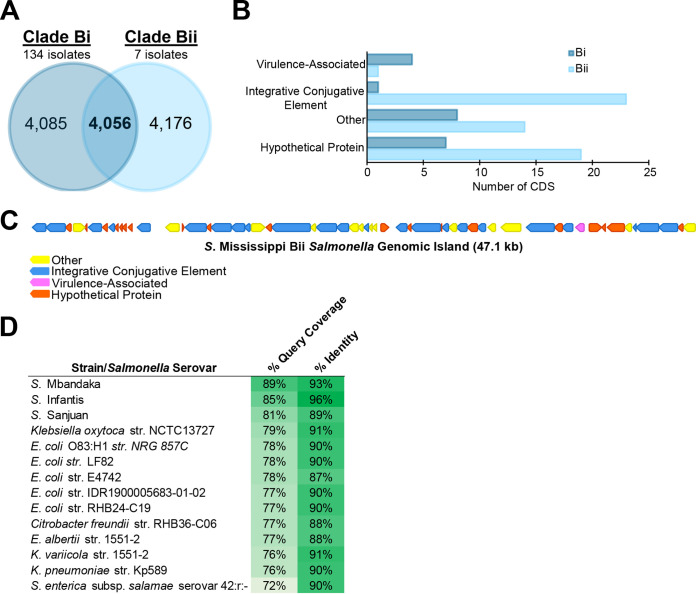
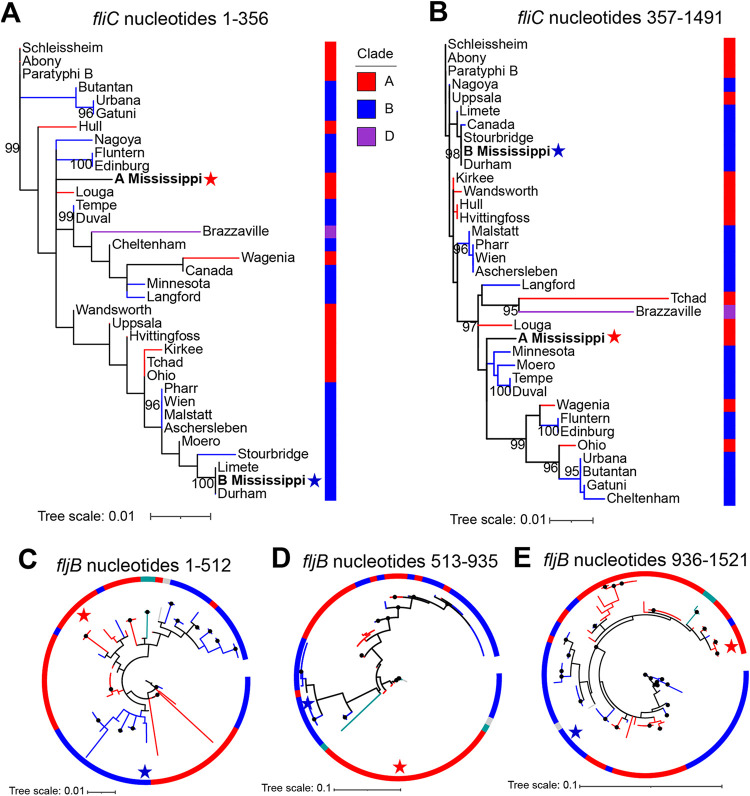
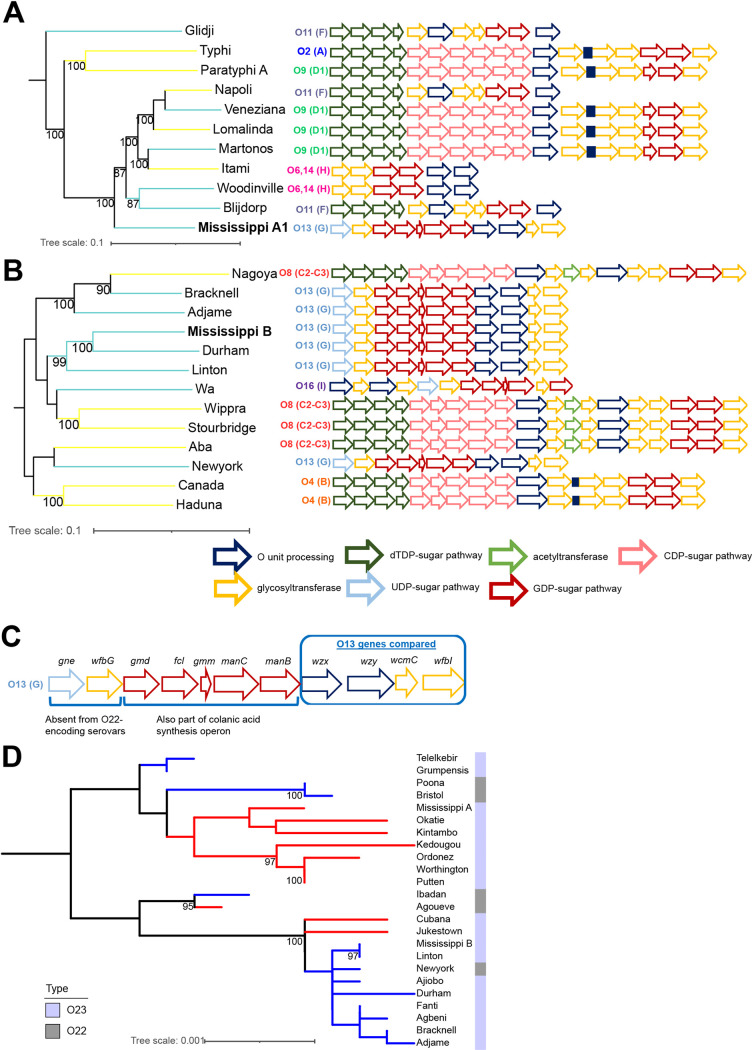
Salmonella enterica serovar Mississippi is the 2nd and 14th leading cause of human clinical salmonellosis in the Australian island state of Tasmania and the United States, respectively. Despite its public health relevance, relatively little is known about this serovar. Comparison of whole-genome sequence (WGS) data of S. Mississippi isolates with WGS data for 317 additional S. enterica serovars placed one clade of S. Mississippi within S. enterica clade B ("clade B Mississippi") and the other within section Typhi in S. enterica clade A ("clade A Mississippi"), suggesting that these clades evolved from different ancestors. Phylogenetic analysis of 364 S. Mississippi isolates from Australia, the United Kingdom, and the United States suggested that the isolates cluster geographically, with U.S. and Australian isolates representing different subclades (Ai and Aii, respectively) within clade A Mississippi and clade B isolates representing the predominant S. Mississippi isolates in the United Kingdom. Intraclade comparisons suggested that different mobile elements, some of which encode virulence factors, are responsible for the observed differences in gene content among isolates within these clades. Specifically, genetic differences among clade A isolates reflect differences in prophage contents, while differences among clade B isolates are due to the acquisition of a 47.1-kb integrative conjugative element (ICE). Phylogenies inferred from antigenic components (fliC, fljB, and O-antigen-processing genes) support that clade A and B Mississippi isolates acquired these loci from different ancestral serovars. Overall, these data support that different S. Mississippi phylogenetic clades are endemic in Australia, the United Kingdom, and the United States. IMPORTANCE The number of known so-called "polyphyletic" serovars (i.e., phylogenetically distinct clades with the same O and H antigenic formulas) continues to increase as additional Salmonella isolates are sequenced. While serotyping remains a valuable tool for reporting and monitoring Salmonella, more discriminatory analyses for classifying polyphyletic serovars may improve surveillance efforts for these serovars, as we found that for S. Mississippi, distinct genotypes predominate at different geographic locations. Our results suggest that the acquisition of genes encoding O and H antigens from different ancestors led to the emergence of two Mississippi clades. Furthermore, our results suggest that different mobile elements contribute to the microevolution and diversification of isolates within these two clades, which has implications for the acquisition of novel adaptations, such as virulence factors.
Keywords: Salmonella; phylogeography; polyphyly; prophage; whole-genome sequencing.
Figures
References
-
- Havelaar AH, Kirk MD, Torgerson PR, Gibb HJ, Hald T, Lake RJ, Praet N, Bellinger DC, de Silva NR, Gargouri N, Speybroeck N, Cawthorne A, Mathers C, Stein C, Angulo FJ, Devleesschauwer B, World Health Organization Foodborne Disease Burden Epidemiology Reference Group . 2015. World Health Organization global estimates and regional comparisons of the burden of foodborne disease in 2010. PLoS Med 12:e1001923. doi: 10.1371/journal.pmed.1001923. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
