

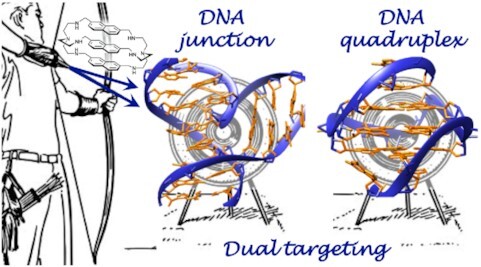
Dual targeting of higher-order DNA structures by azacryptands induces DNA junction-mediated DNA damage in cancer cells
- PMID: 34551430
- PMCID: PMC8501980
- DOI: 10.1093/nar/gkab796
Dual targeting of higher-order DNA structures by azacryptands induces DNA junction-mediated DNA damage in cancer cells
Abstract
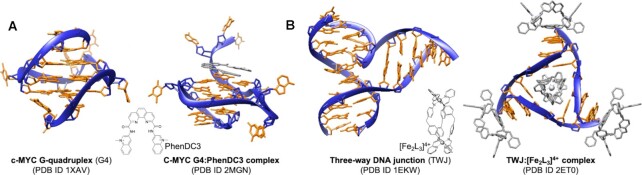
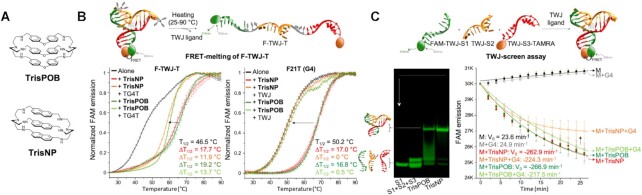
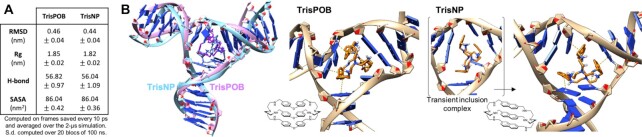
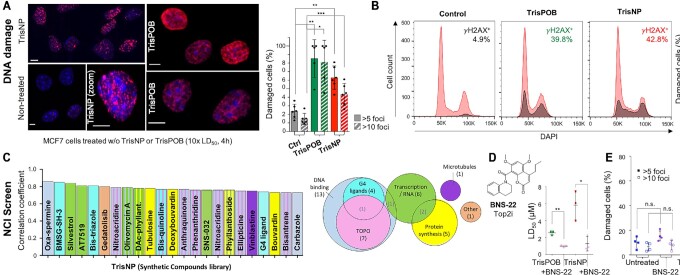
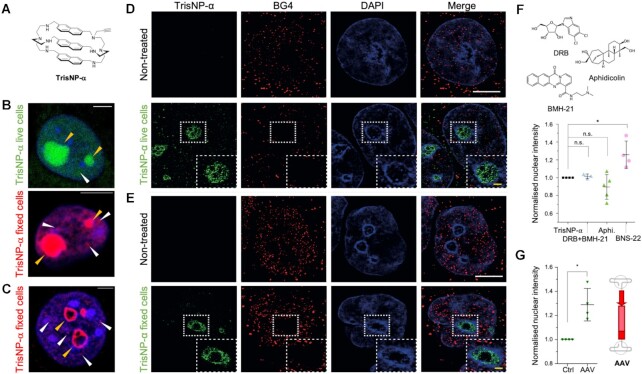
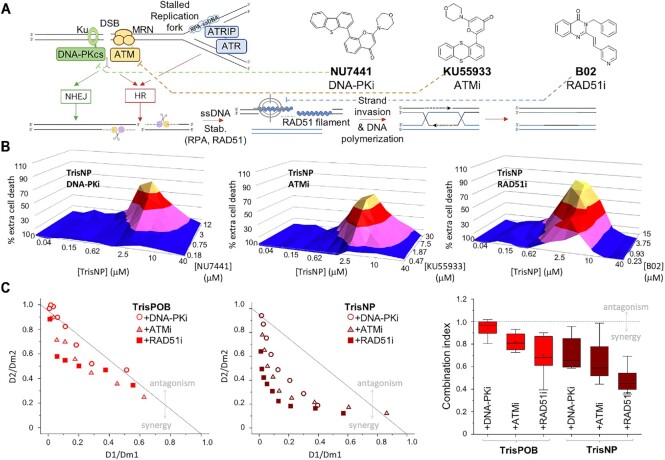
DNA is intrinsically dynamic and folds transiently into alternative higher-order structures such as G-quadruplexes (G4s) and three-way DNA junctions (TWJs). G4s and TWJs can be stabilised by small molecules (ligands) that have high chemotherapeutic potential, either as standalone DNA damaging agents or combined in synthetic lethality strategies. While previous approaches have claimed to use ligands that specifically target either G4s or TWJs, we report here on a new approach in which ligands targeting both TWJs and G4s in vitro demonstrate cellular effects distinct from that of G4 ligands, and attributable to TWJ targeting. The DNA binding modes of these new, dual TWJ-/G4-ligands were studied by a panel of in vitro methods and theoretical simulations, and their cellular properties by extensive cell-based assays. We show here that cytotoxic activity of TWJ-/G4-ligands is mitigated by the DNA damage response (DDR) and DNA topoisomerase 2 (TOP2), making them different from typical G4-ligands, and implying a pivotal role of TWJs in cells. We designed and used a clickable ligand, TrisNP-α, to provide unique insights into the TWJ landscape in cells and its modulation upon co-treatments. This wealth of data was exploited to design an efficient synthetic lethality strategy combining dual ligands with clinically relevant DDR inhibitors.
© The Author(s) 2021. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Hustedt N., Durocher D.. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2017; 19:1–9. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
