

Genetic and epigenetic coordination of cortical interneuron development
- PMID: 34552240
- PMCID: PMC9316417
- DOI: 10.1038/s41586-021-03933-1
Genetic and epigenetic coordination of cortical interneuron development
Abstract
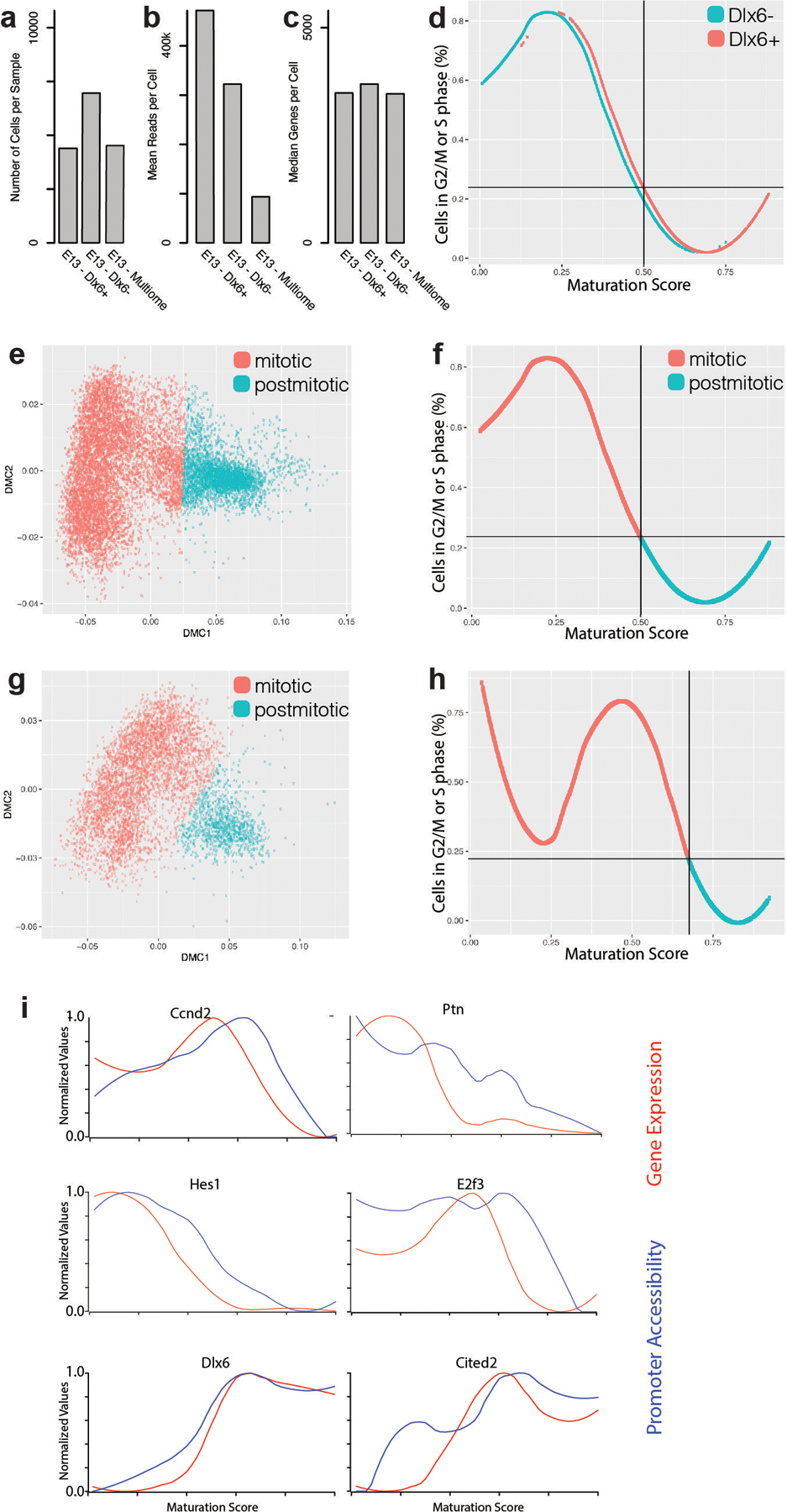
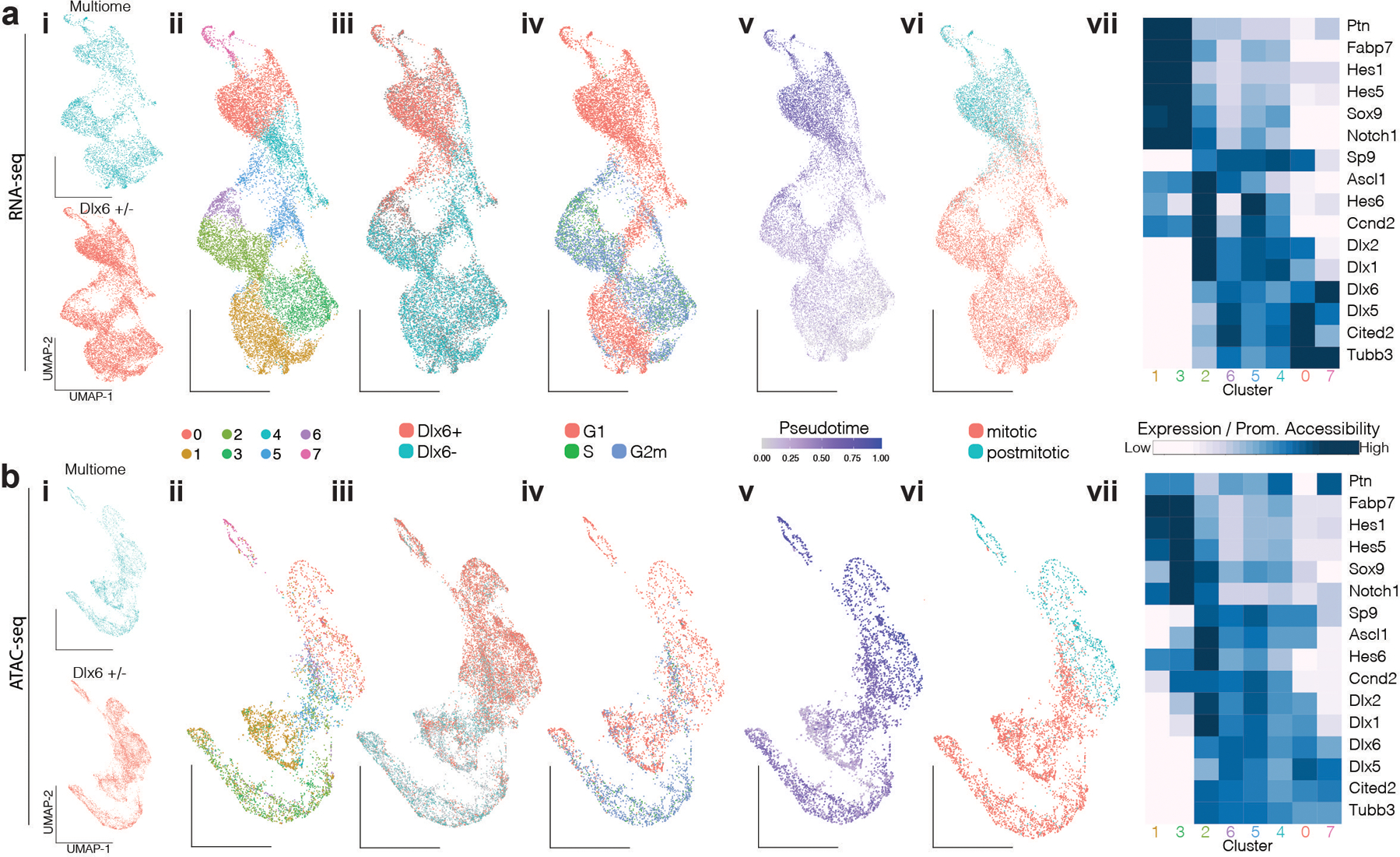
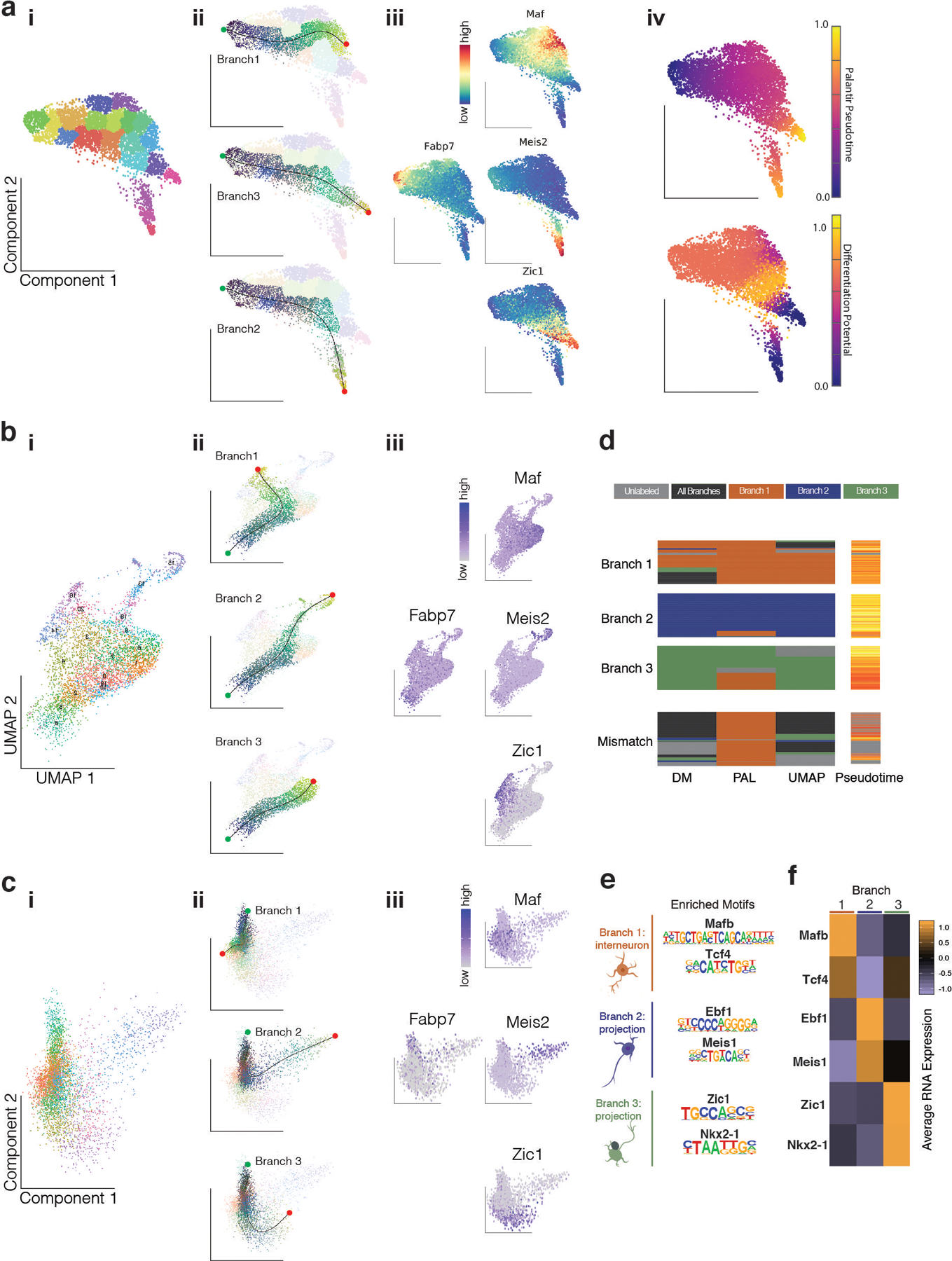
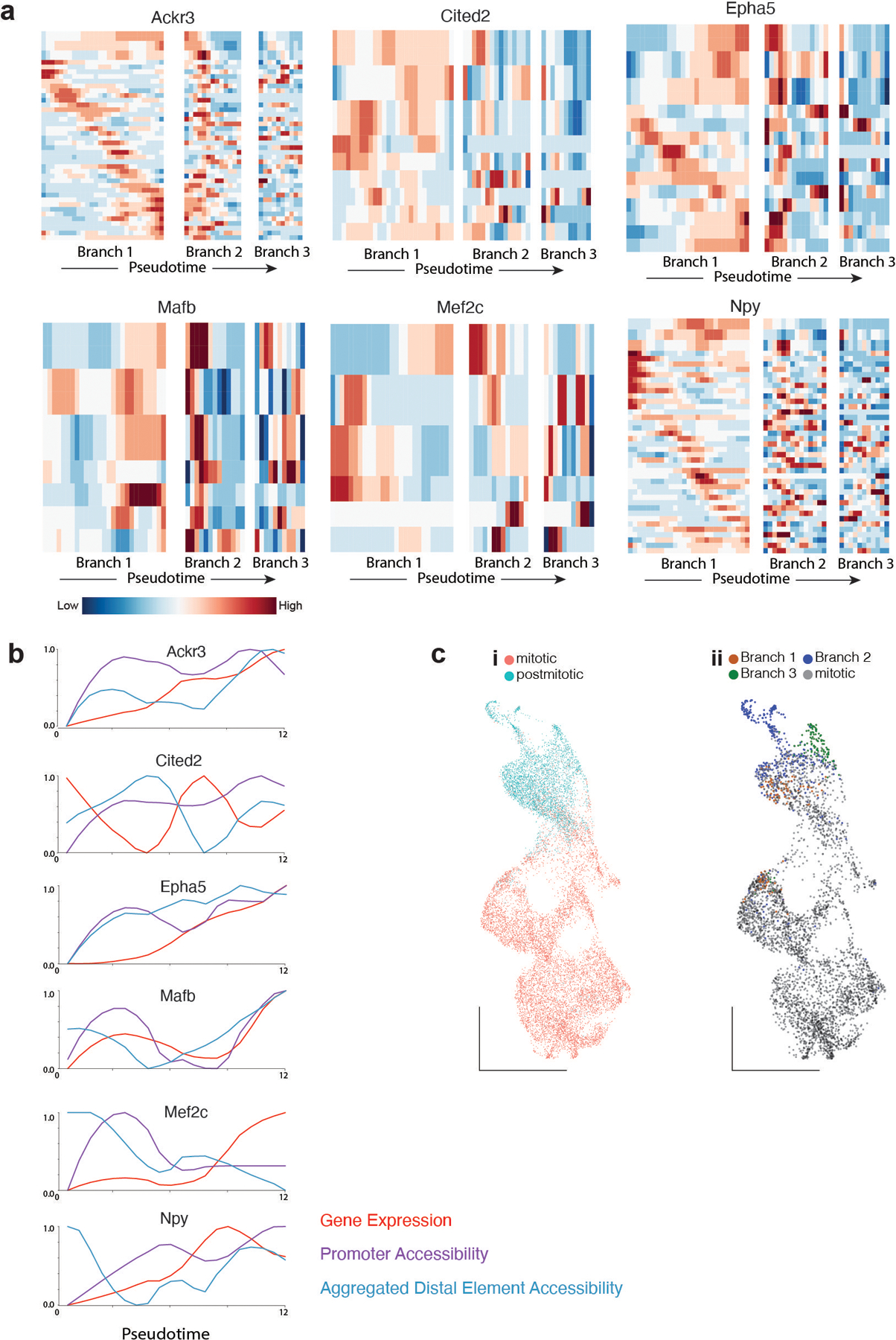
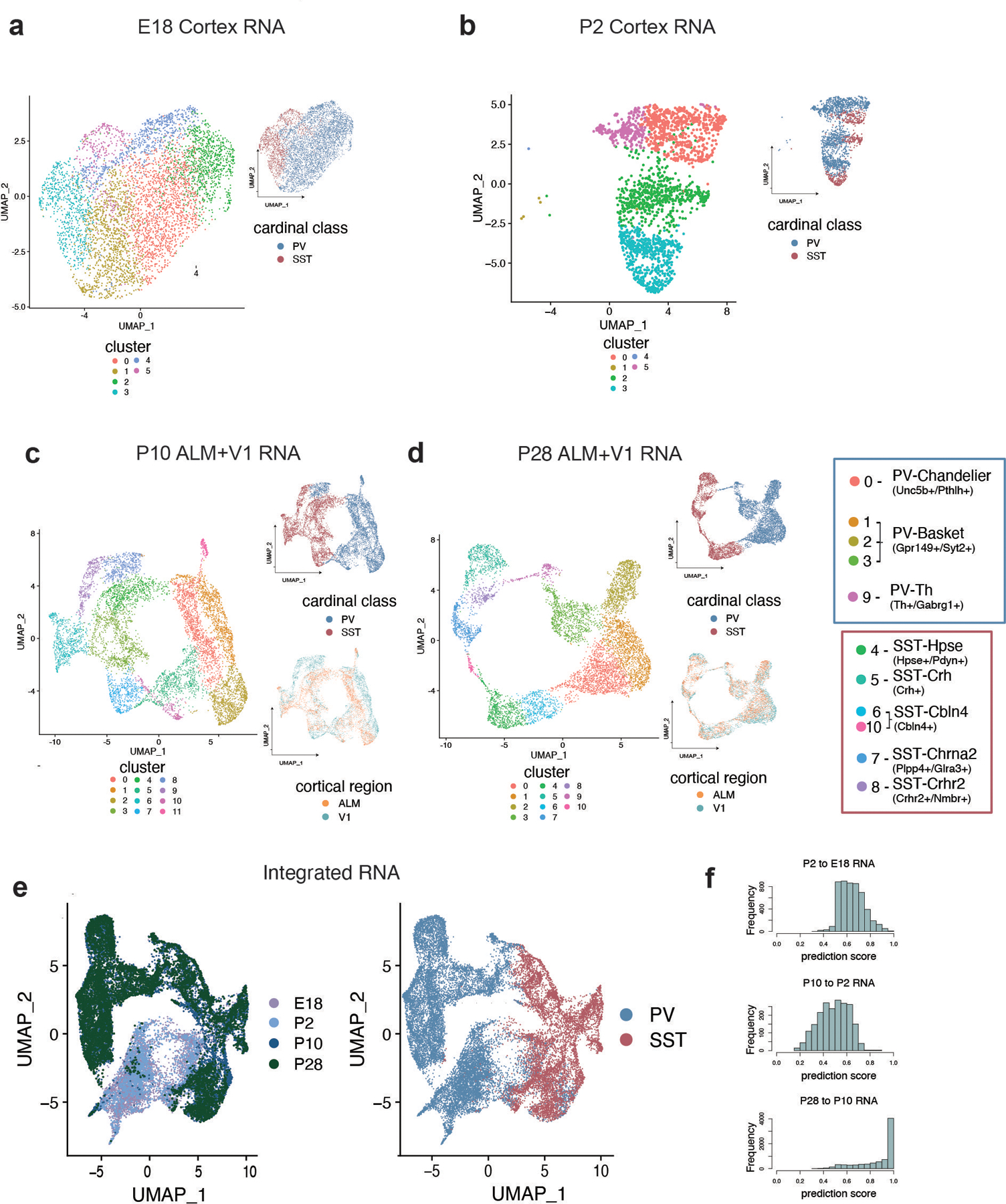
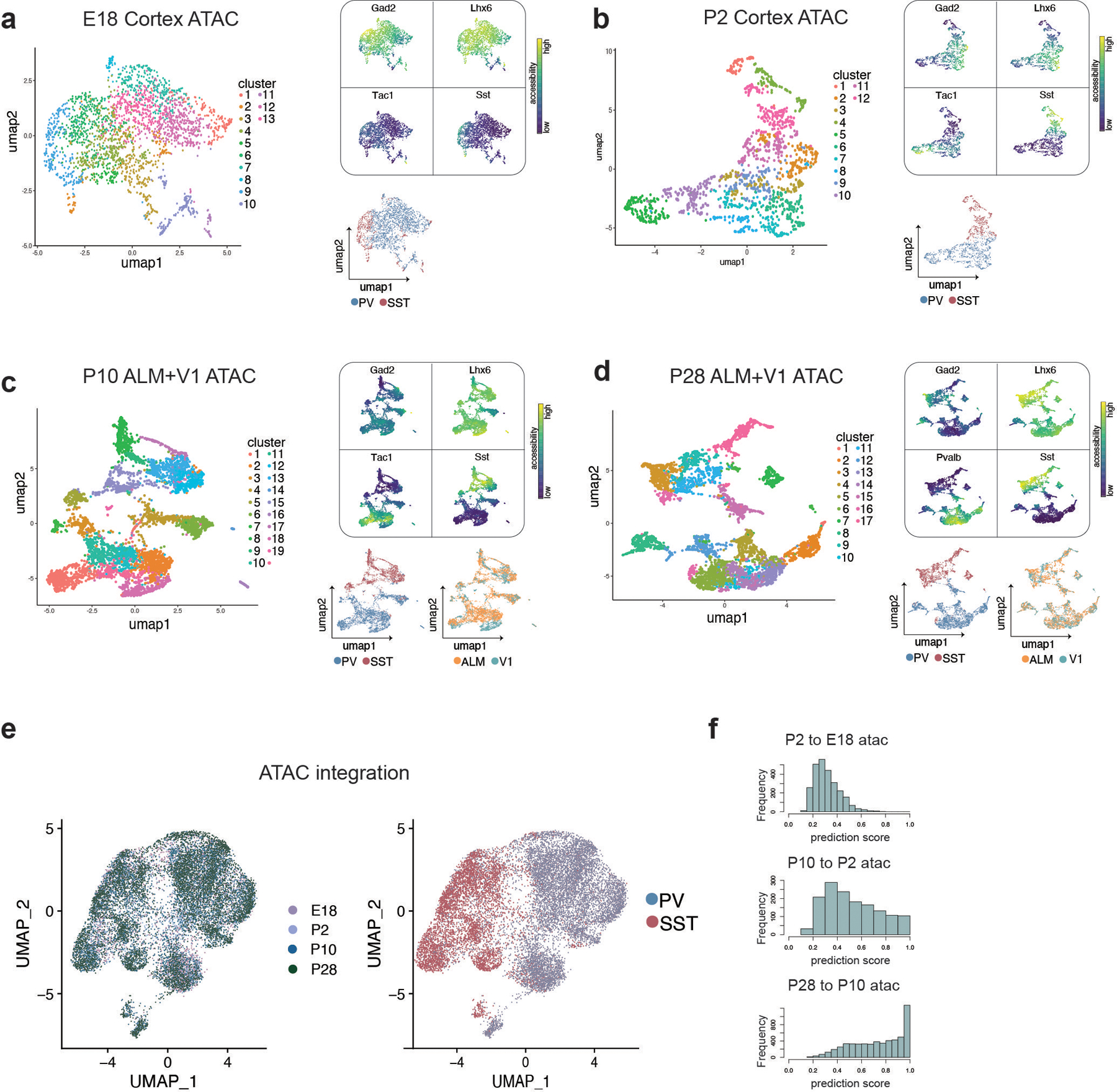
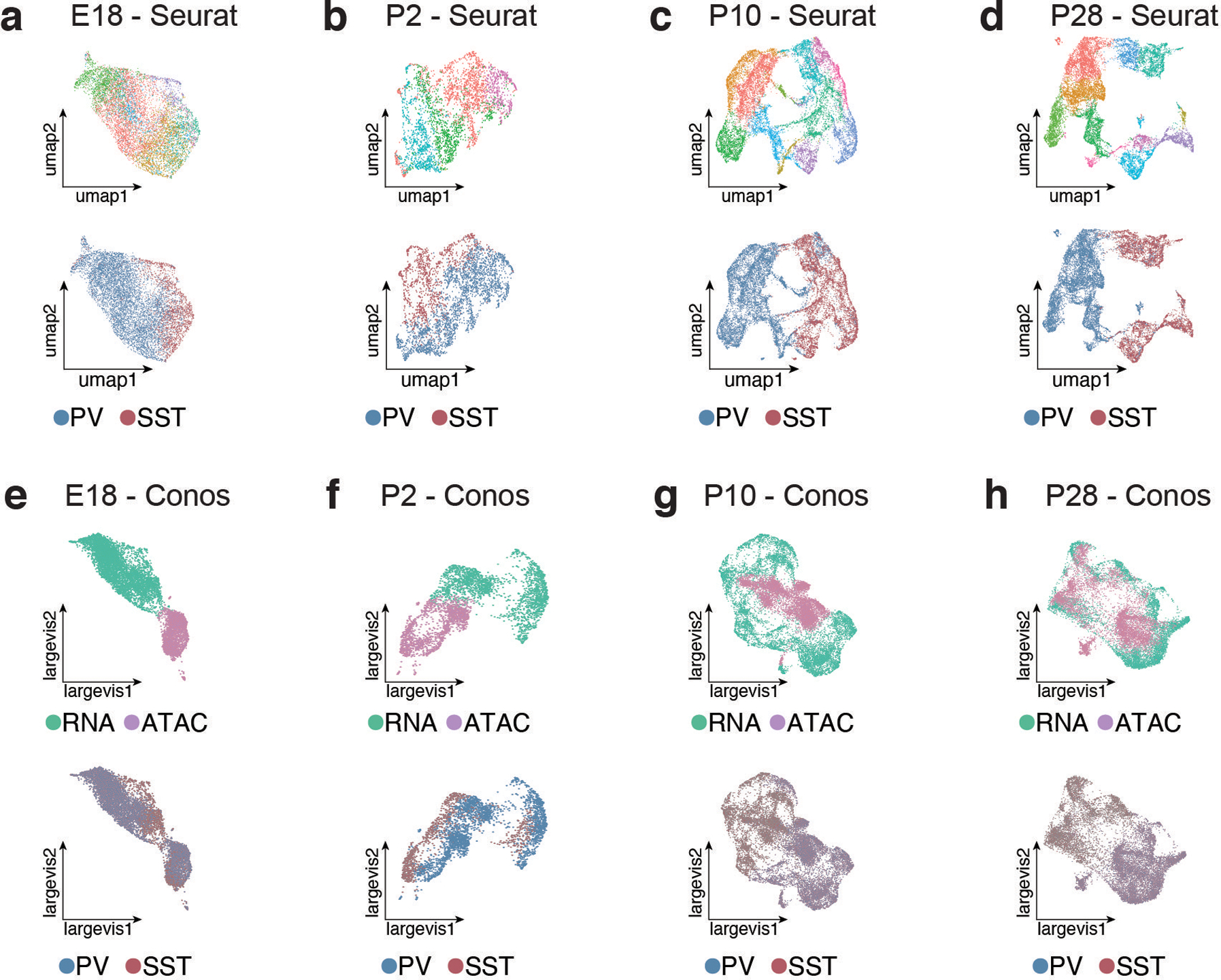
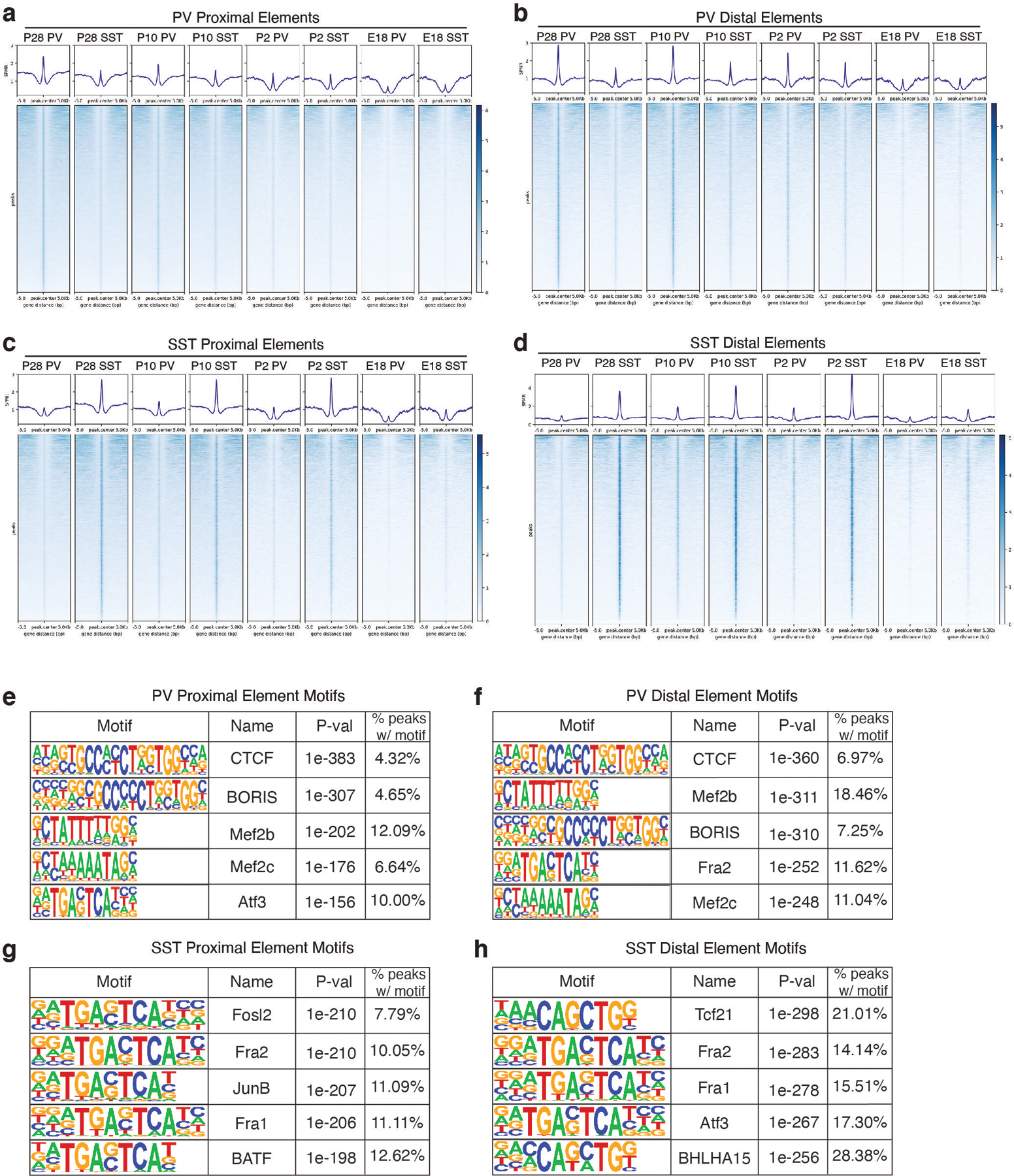
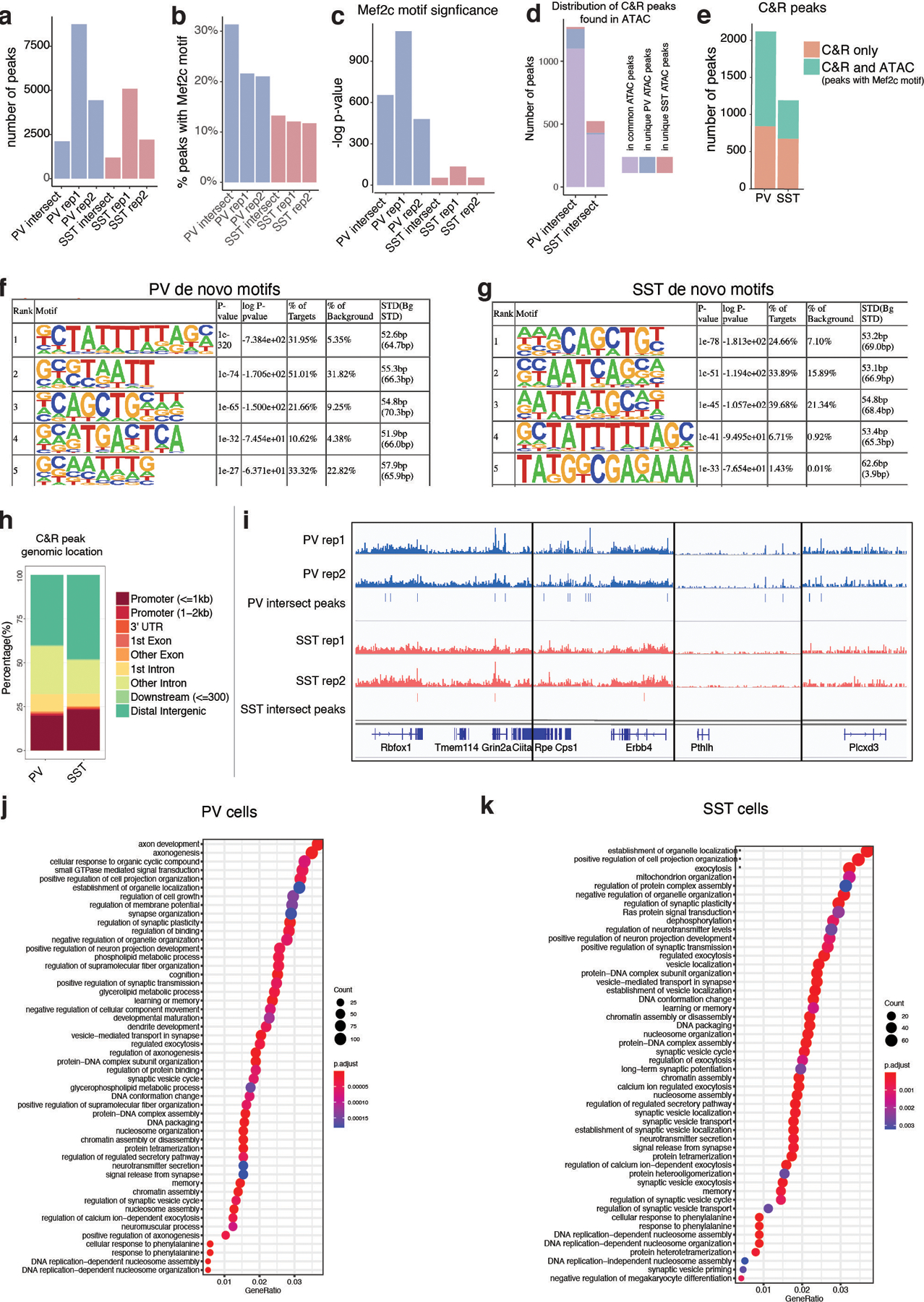
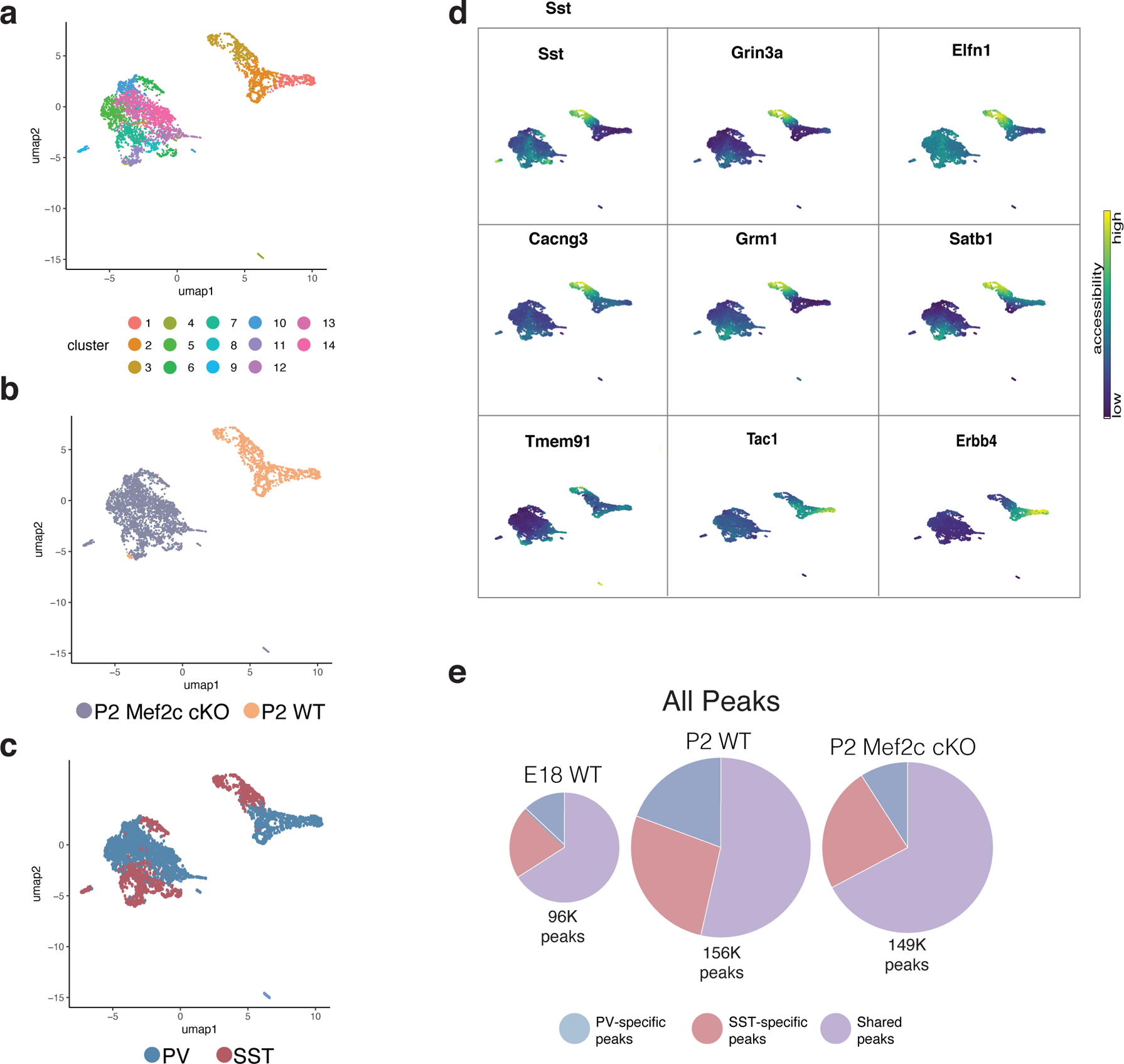
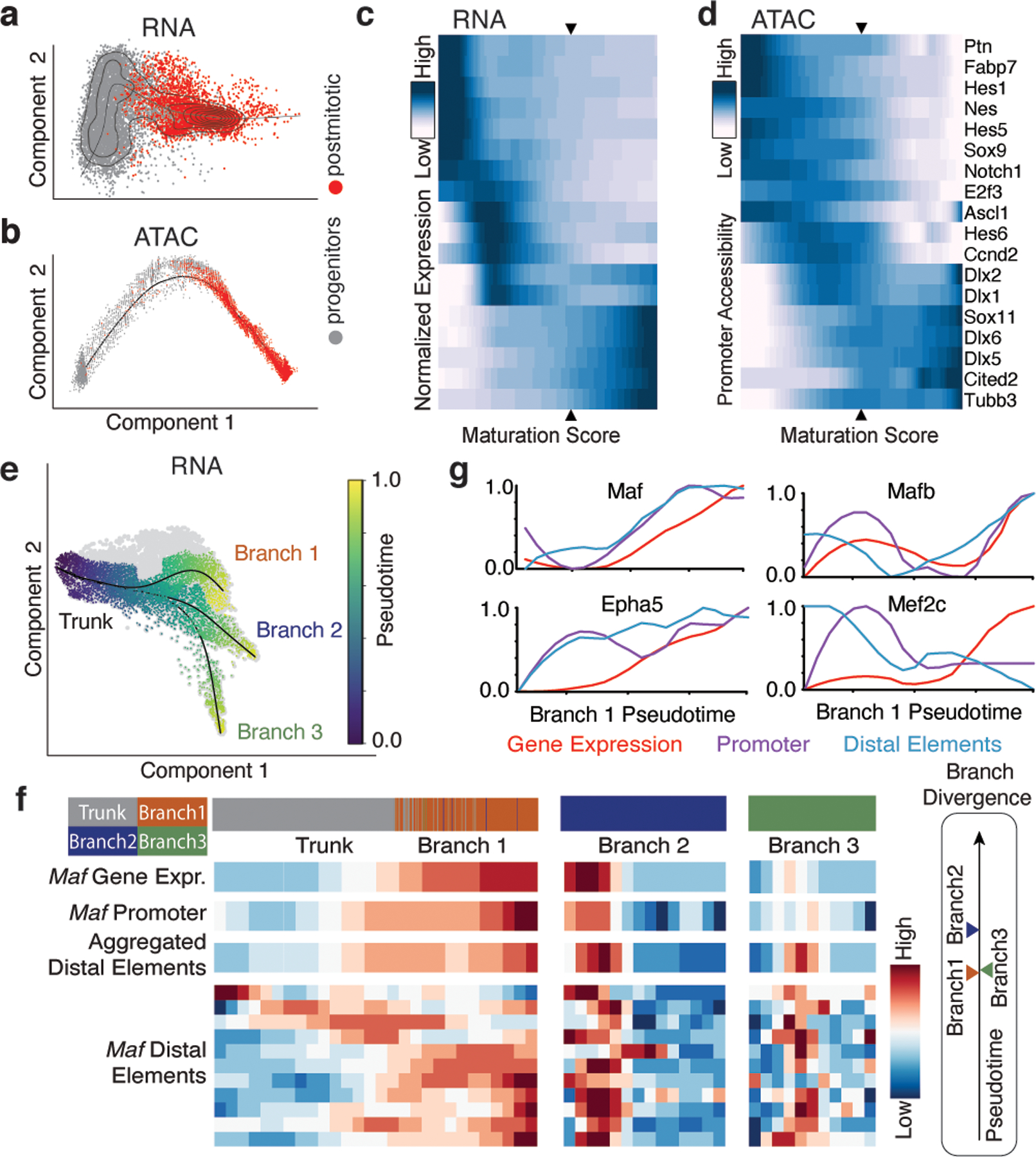
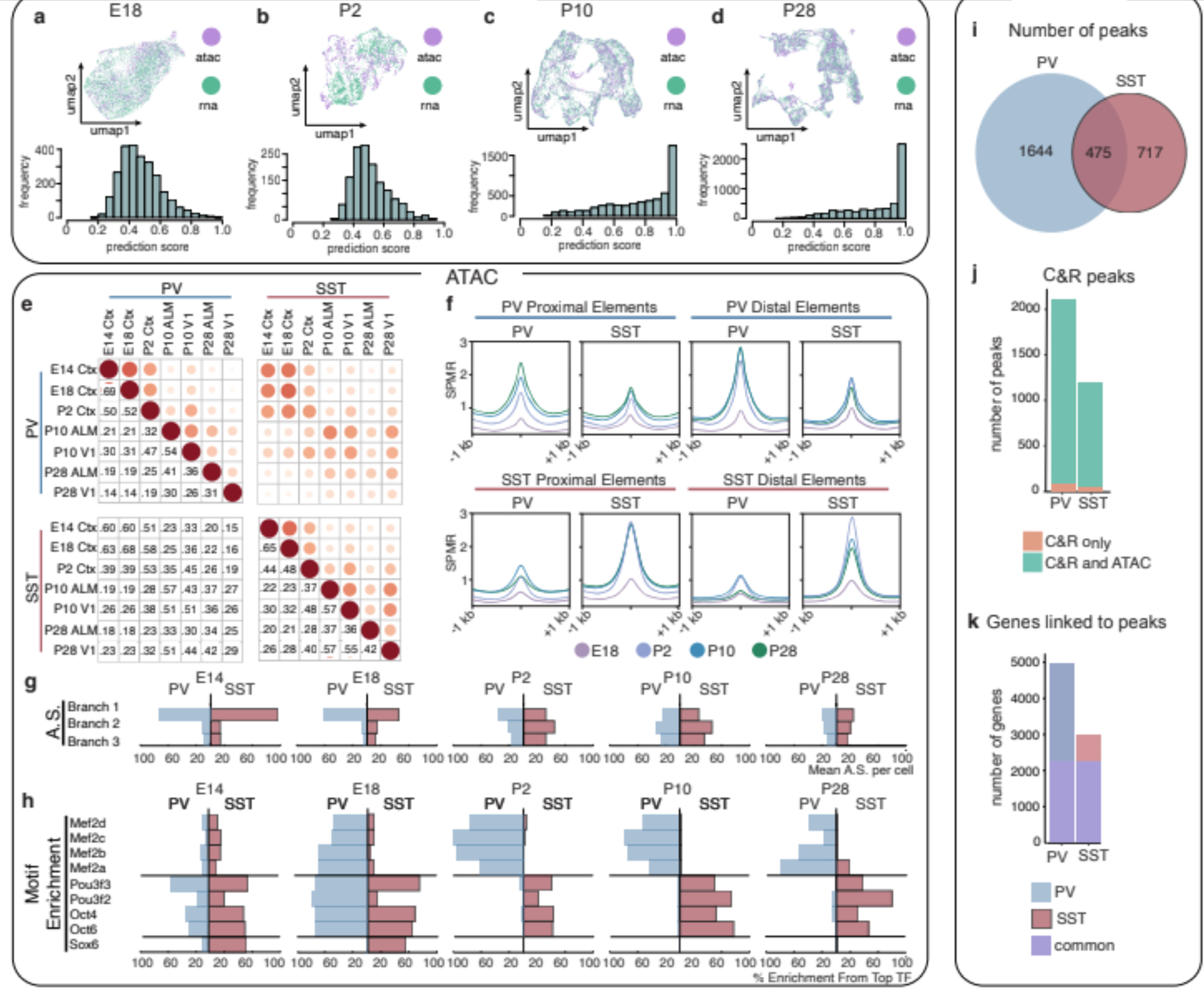
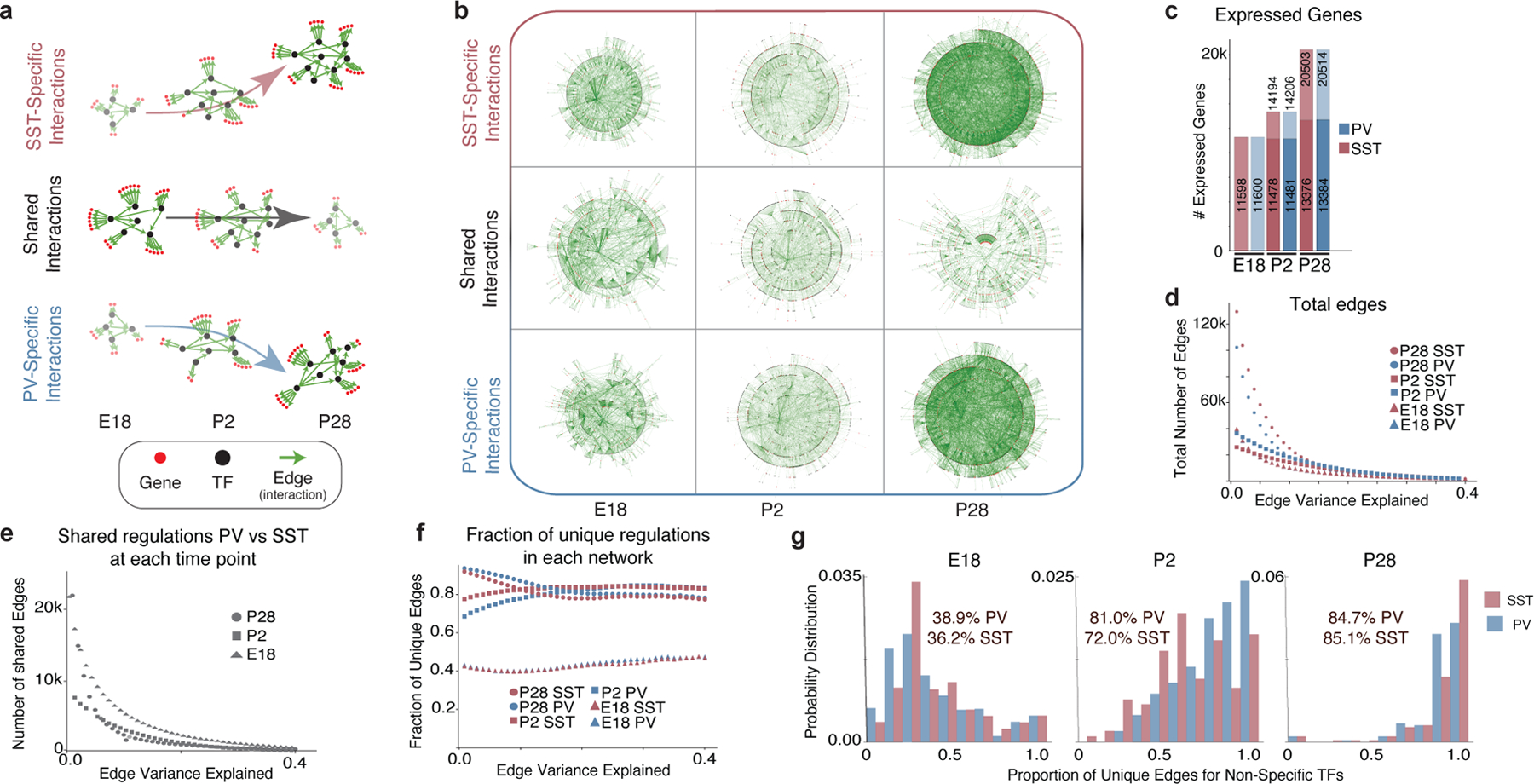
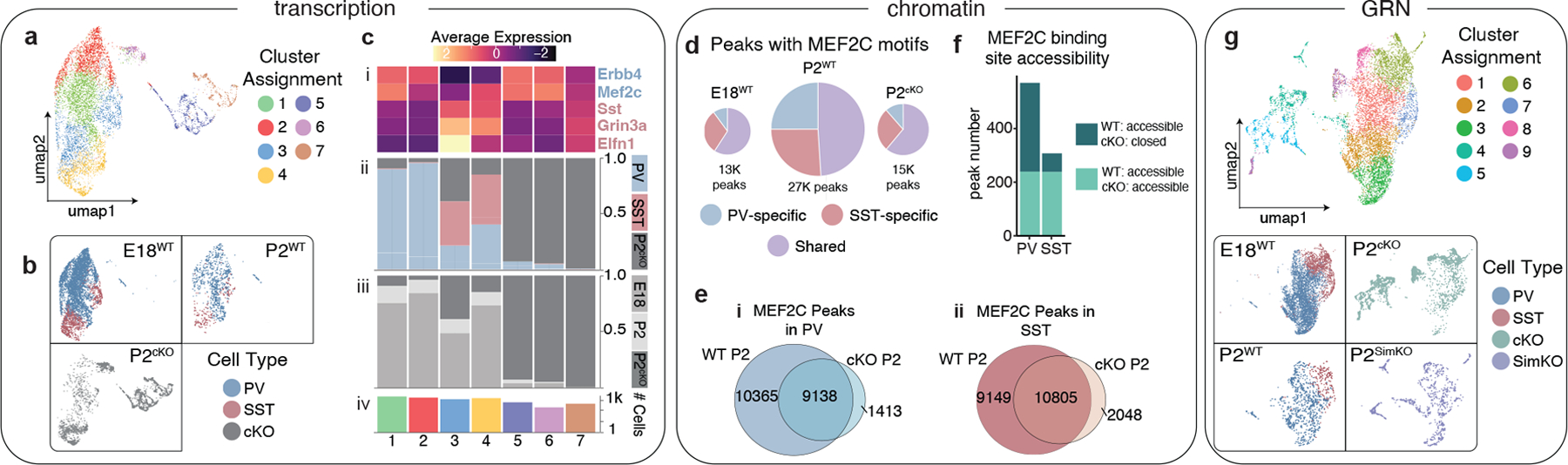
One of the hallmarks of the cerebral cortex is the extreme diversity of interneurons1-3. The two largest subtypes of cortical interneurons, parvalbumin- and somatostatin-positive cells, are morphologically and functionally distinct in adulthood but arise from common lineages within the medial ganglionic eminence4-11. This makes them an attractive model for studying the generation of cell diversity. Here we examine how developmental changes in transcription and chromatin structure enable these cells to acquire distinct identities in the mouse cortex. Generic interneuron features are first detected upon cell cycle exit through the opening of chromatin at distal elements. By constructing cell-type-specific gene regulatory networks, we observed that parvalbumin- and somatostatin-positive cells initiate distinct programs upon settling within the cortex. We used these networks to model the differential transcriptional requirement of a shared regulator, Mef2c, and confirmed the accuracy of our predictions through experimental loss-of-function experiments. We therefore reveal how a common molecular program diverges to enable these neuronal subtypes to acquire highly specialized properties by adulthood. Our methods provide a framework for examining the emergence of cellular diversity, as well as for quantifying and predicting the effect of candidate genes on cell-type-specific development.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing Interests
The authors declare no competing interests. Correspondence and requests for materials should be addressed to G.F. (
Figures
References
Methods References
-
- Vong LH, Ragusa MJ & Schwarz JJ Generation of conditional Mef2cloxp/loxp mice for temporal- and tissue-specific analyses. Genesis 43, 43–48 (2005). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
