

Biologically informed deep neural network for prostate cancer discovery
- PMID: 34552244
- PMCID: PMC8514339
- DOI: 10.1038/s41586-021-03922-4
Biologically informed deep neural network for prostate cancer discovery
Abstract
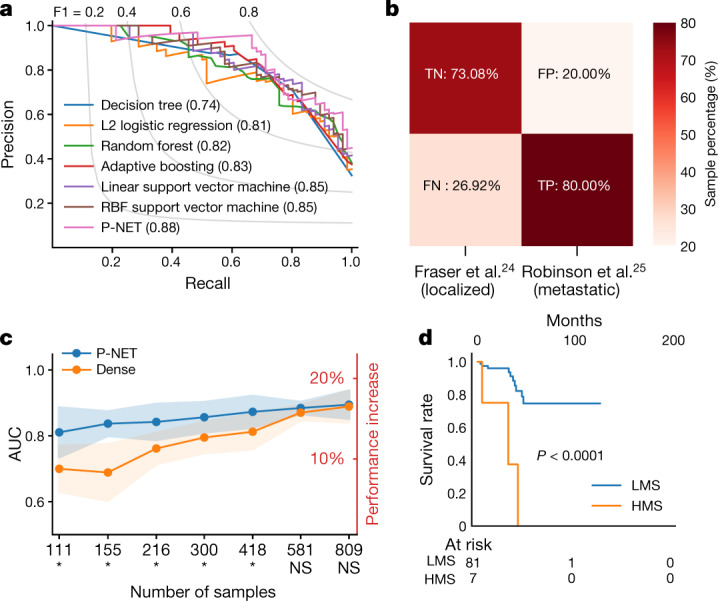
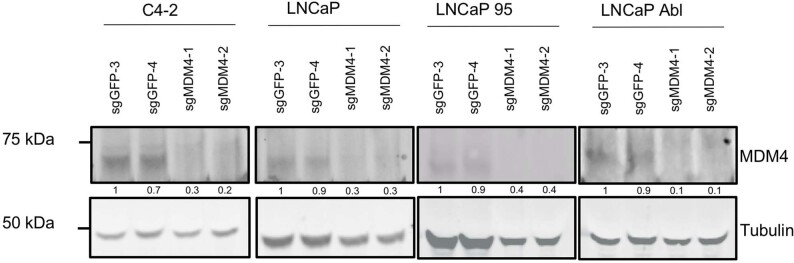
The determination of molecular features that mediate clinically aggressive phenotypes in prostate cancer remains a major biological and clinical challenge1,2. Recent advances in interpretability of machine learning models as applied to biomedical problems may enable discovery and prediction in clinical cancer genomics3-5. Here we developed P-NET-a biologically informed deep learning model-to stratify patients with prostate cancer by treatment-resistance state and evaluate molecular drivers of treatment resistance for therapeutic targeting through complete model interpretability. We demonstrate that P-NET can predict cancer state using molecular data with a performance that is superior to other modelling approaches. Moreover, the biological interpretability within P-NET revealed established and novel molecularly altered candidates, such as MDM4 and FGFR1, which were implicated in predicting advanced disease and validated in vitro. Broadly, biologically informed fully interpretable neural networks enable preclinical discovery and clinical prediction in prostate cancer and may have general applicability across cancer types.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
W.C.H. is a consultant for Thermo Fisher, Solasta Ventures, iTeos, Frontier Medicines, Tyra Biosciences, MPM Capital, KSQ Therapeutics and Parexel and is a founder of KSQ Therapeutics. E.M.V. is a consultant/advisor for Tango Therapeutics, Genome Medical, Invitae, Enara Bio, Janssen, Manifold Bio and Monte Rosa Therapeutics. E.M.V. receives research support from Novartis and BMS. The other authors declare no competing interests.
Figures











Comment in
-
Is Artificial Intelligence Ready for "Primetime" in Urology?BJU Int. 2021 Dec;128(6):659-660. doi: 10.1111/bju.15633. BJU Int. 2021. PMID: 34856063 No abstract available.
-
Uro-Science.J Urol. 2022 Aug;208(2):468-469. doi: 10.1097/JU.0000000000002770. Epub 2022 May 20. J Urol. 2022. PMID: 35593059 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
