

NSD2 dimethylation at H3K36 promotes lung adenocarcinoma pathogenesis
- PMID: 34555356
- PMCID: PMC8571016
- DOI: 10.1016/j.molcel.2021.08.034
NSD2 dimethylation at H3K36 promotes lung adenocarcinoma pathogenesis
Abstract
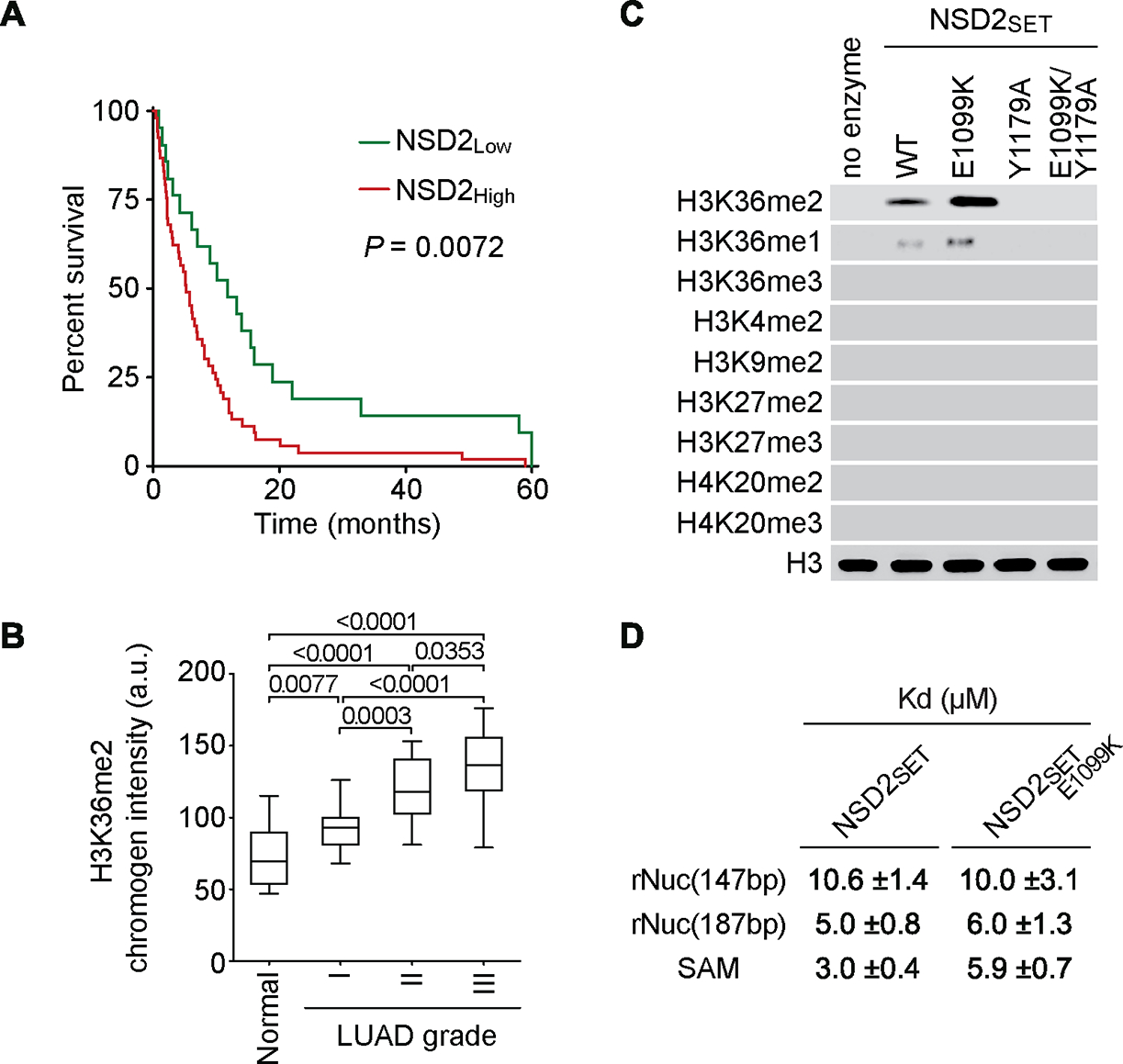
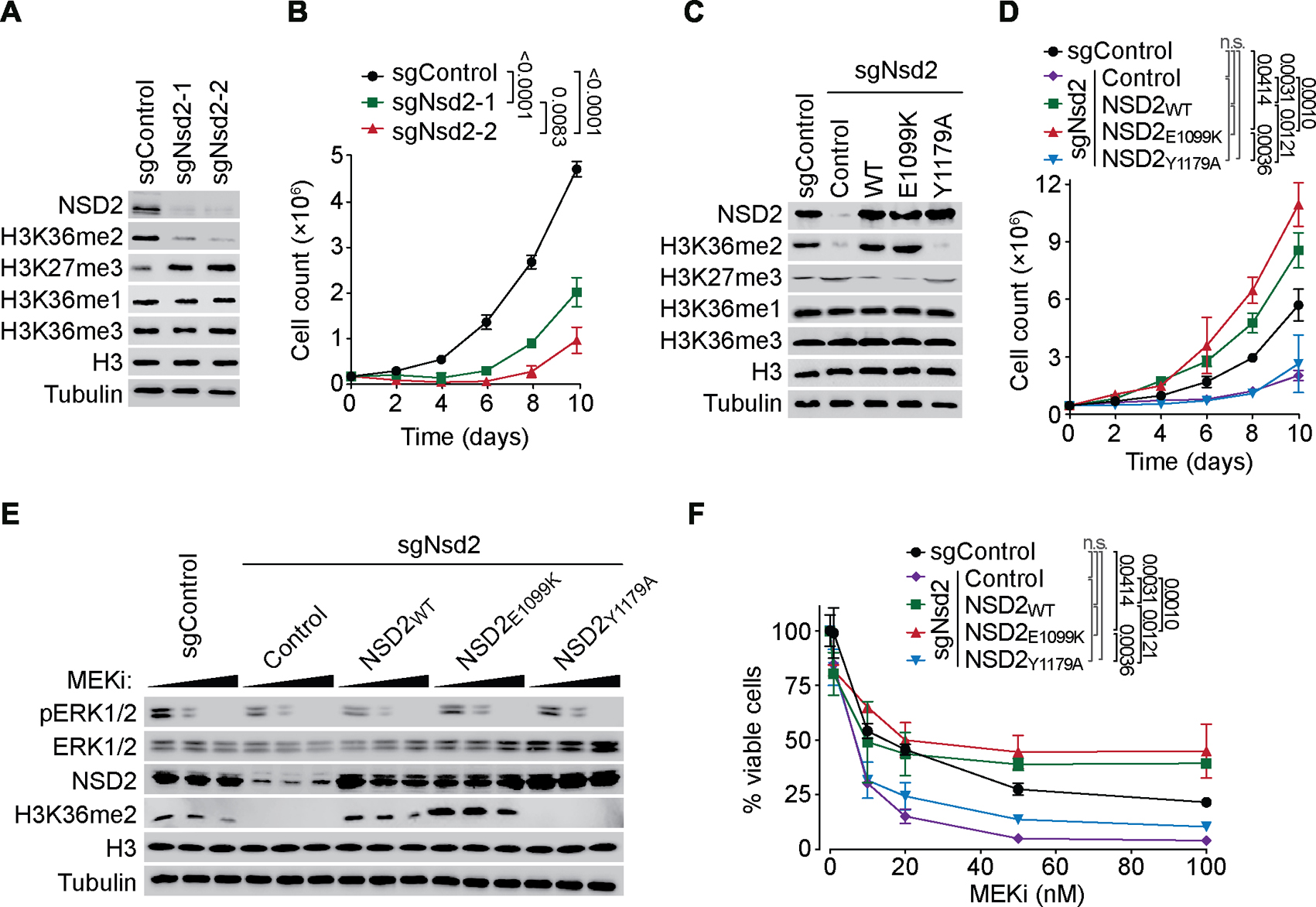
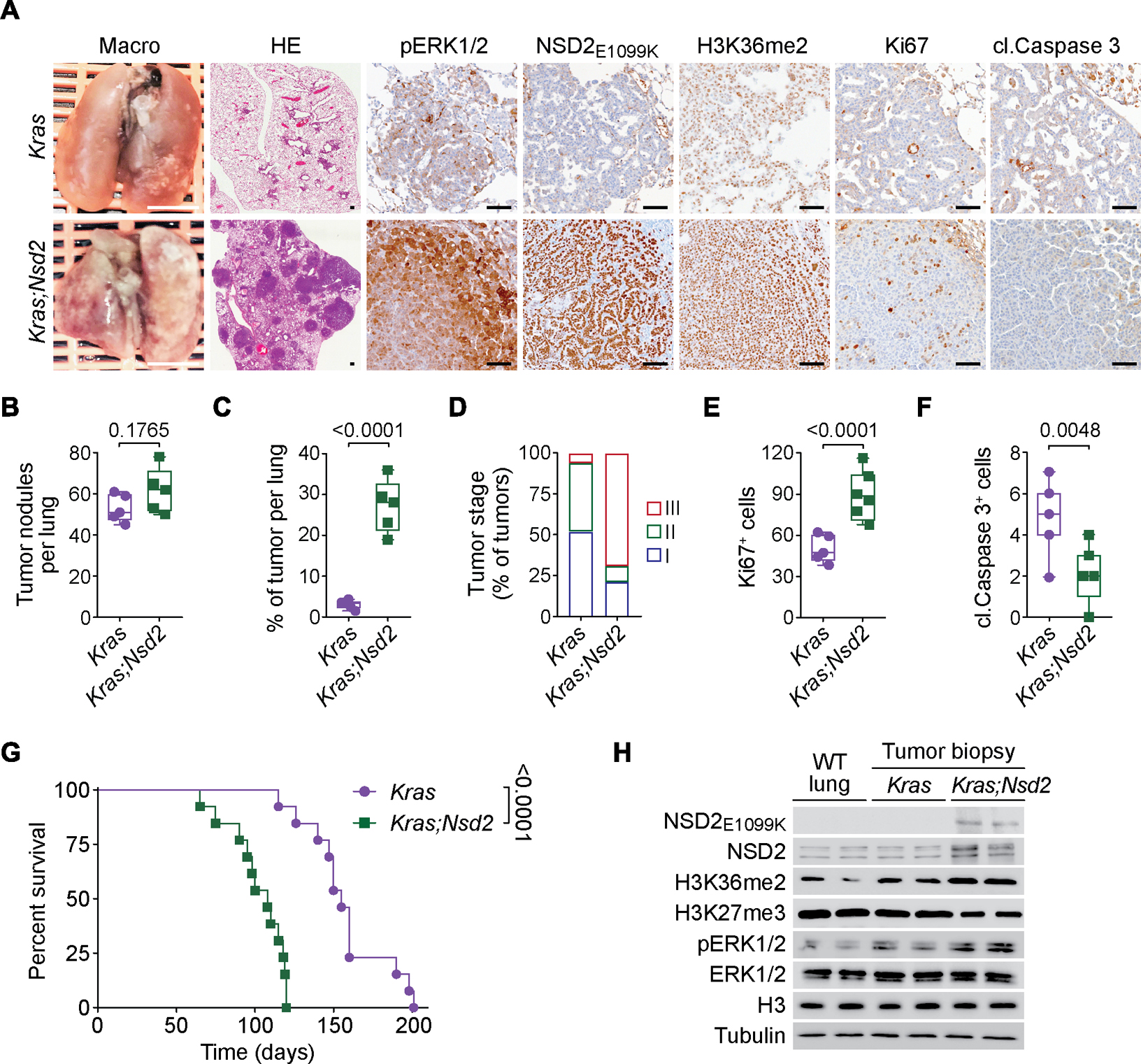
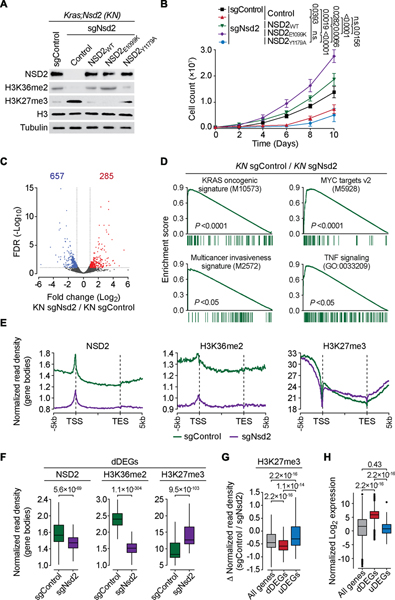
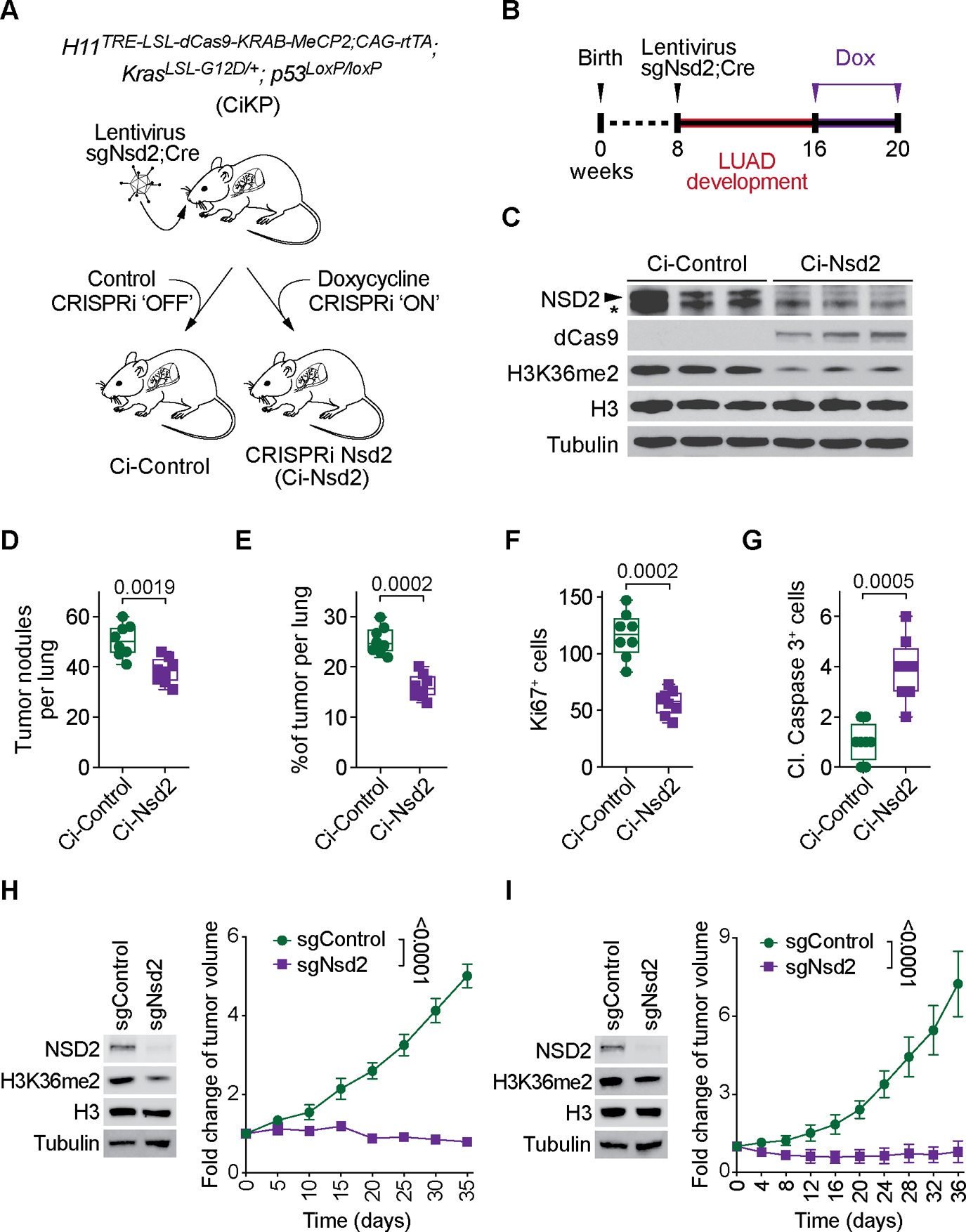
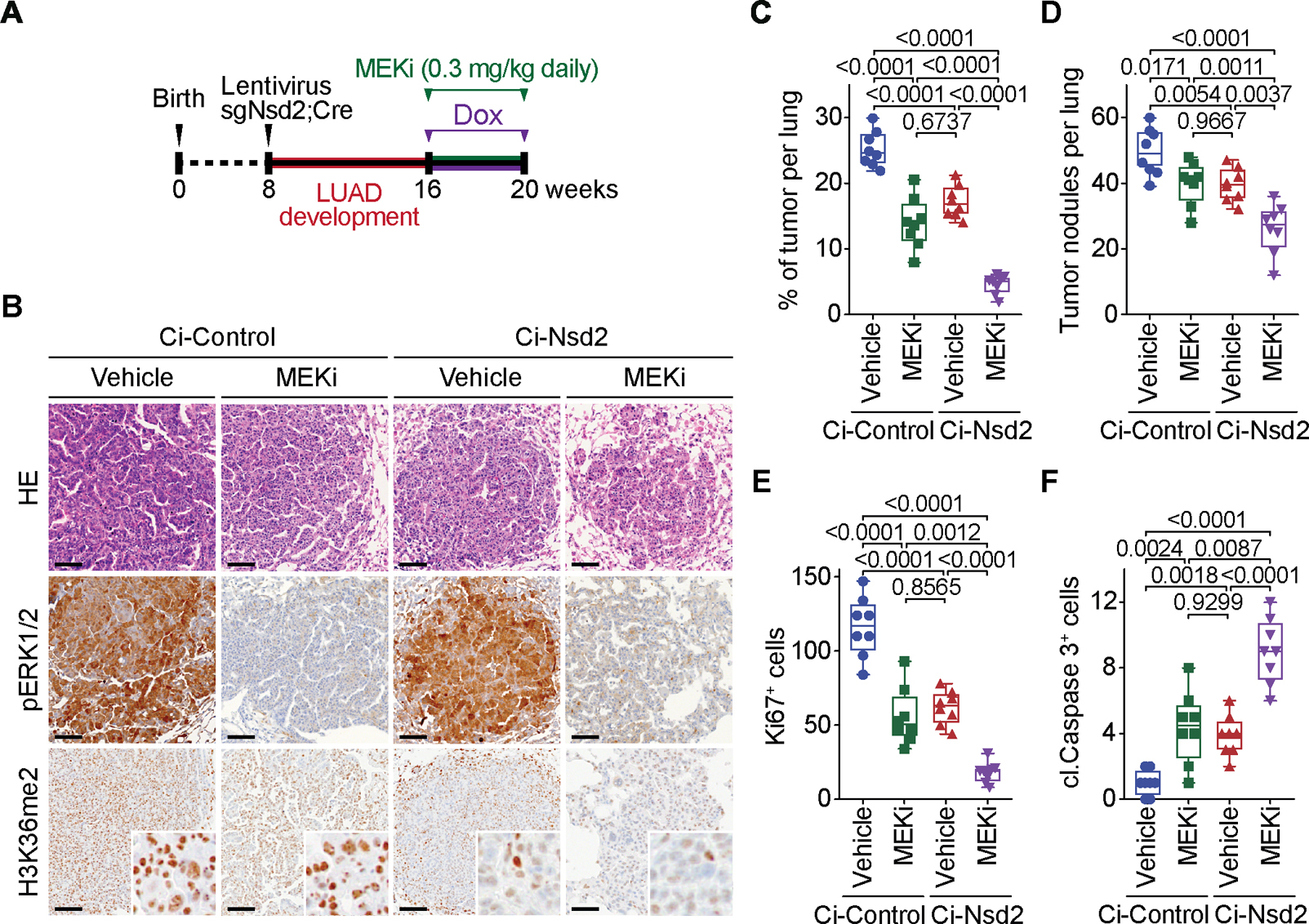
The etiological role of NSD2 enzymatic activity in solid tumors is unclear. Here we show that NSD2, via H3K36me2 catalysis, cooperates with oncogenic KRAS signaling to drive lung adenocarcinoma (LUAD) pathogenesis. In vivo expression of NSD2E1099K, a hyperactive variant detected in individuals with LUAD, rapidly accelerates malignant tumor progression while decreasing survival in KRAS-driven LUAD mouse models. Pathologic H3K36me2 generation by NSD2 amplifies transcriptional output of KRAS and several complementary oncogenic gene expression programs. We establish a versatile in vivo CRISPRi-based system to test gene functions in LUAD and find that NSD2 loss strongly attenuates tumor progression. NSD2 knockdown also blocks neoplastic growth of PDXs (patient-dervived xenografts) from primary LUAD. Finally, a treatment regimen combining NSD2 depletion with MEK1/2 inhibition causes nearly complete regression of LUAD tumors. Our work identifies NSD2 as a bona fide LUAD therapeutic target and suggests a pivotal epigenetic role of the NSD2-H3K36me2 axis in sustaining oncogenic signaling.
Keywords: CRISPR interference mouse model; H3K36; KRAS; MEK inhibition; NSD2; chromatin; epigenetics; histone methylation; lung adenocarcinoma; lung cancer.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests O.G. is a co-scientific founder, consultant, and stockholder of EpiCypher, Inc. and K36 Therapeutics, Inc. P.K.M. is a scientific co-founder, consultant, and stockholder of Amplified Medicines, Inc. and Ikena Oncology, Inc.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
