

Mutations in Noncoding Cis-Regulatory Elements Reveal Cancer Driver Cistromes in Luminal Breast Cancer
- PMID: 34556523
- PMCID: PMC9398156
- DOI: 10.1158/1541-7786.MCR-21-0471
Mutations in Noncoding Cis-Regulatory Elements Reveal Cancer Driver Cistromes in Luminal Breast Cancer
Abstract
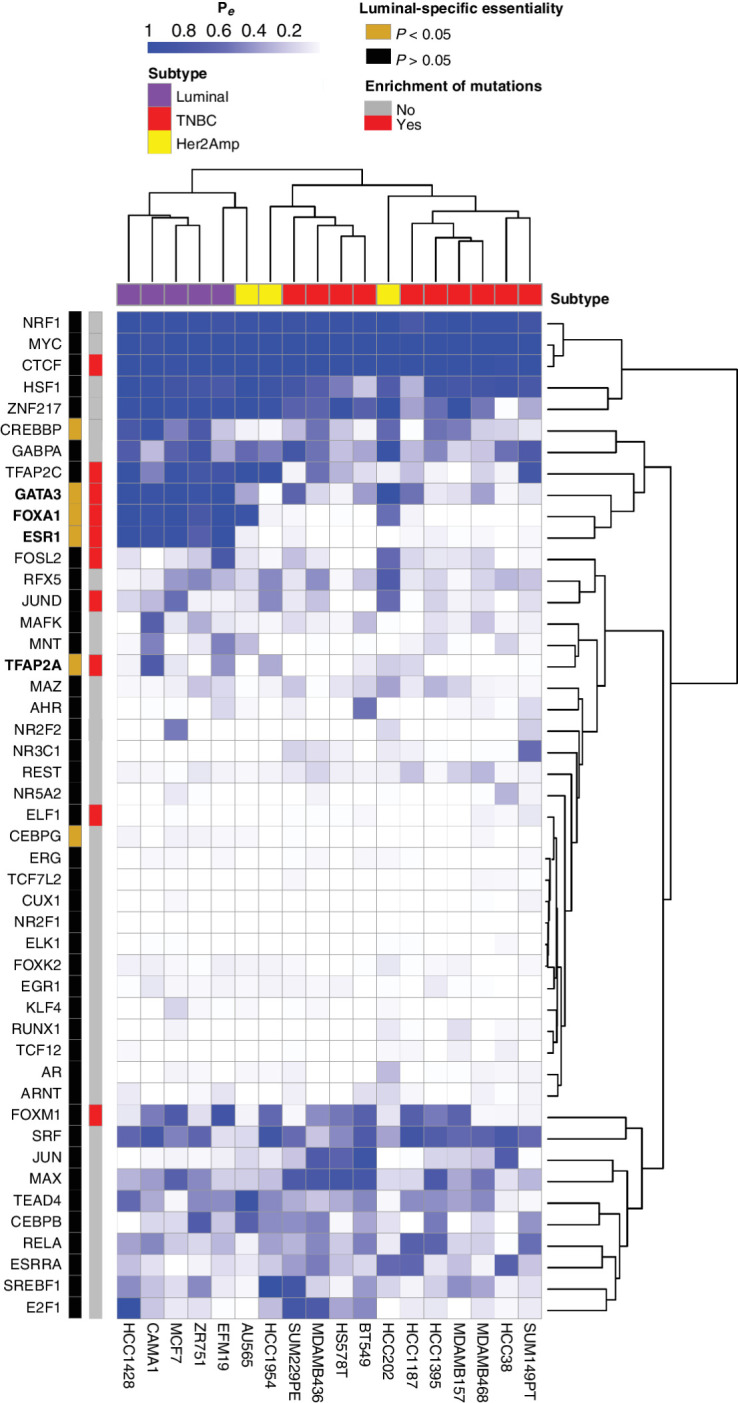
Whole-genome sequencing of primary breast tumors enabled the identification of cancer driver genes and noncoding cancer driver plexuses from somatic mutations. However, differentiating driver from passenger events among noncoding genetic variants remains a challenge. Herein, we reveal cancer-driver cis-regulatory elements linked to transcription factors previously shown to be involved in development of luminal breast cancers by defining a tumor-enriched catalogue of approximately 100,000 unique cis-regulatory elements from 26 primary luminal estrogen receptor (ER)+ progesterone receptor (PR)+ breast tumors. Integrating this catalog with somatic mutations from 350 publicly available breast tumor whole genomes, we uncovered cancer driver cistromes, defined as the sum of binding sites for a transcription factor, for ten transcription factors in luminal breast cancer such as FOXA1 and ER, nine of which are essential for growth in breast cancer with four exclusive to the luminal subtype. Collectively, we present a strategy to find cancer driver cistromes relying on quantifying the enrichment of noncoding mutations over cis-regulatory elements concatenated into a functional unit. IMPLICATIONS: Mapping the accessible chromatin of luminal breast cancer led to discovery of an accumulation of mutations within cistromes of transcription factors essential to luminal breast cancer. This demonstrates coopting of regulatory networks to drive cancer and provides a framework to derive insight into the noncoding space of cancer.
©2021 The Authors; Published by the American Association for Cancer Research.
Figures
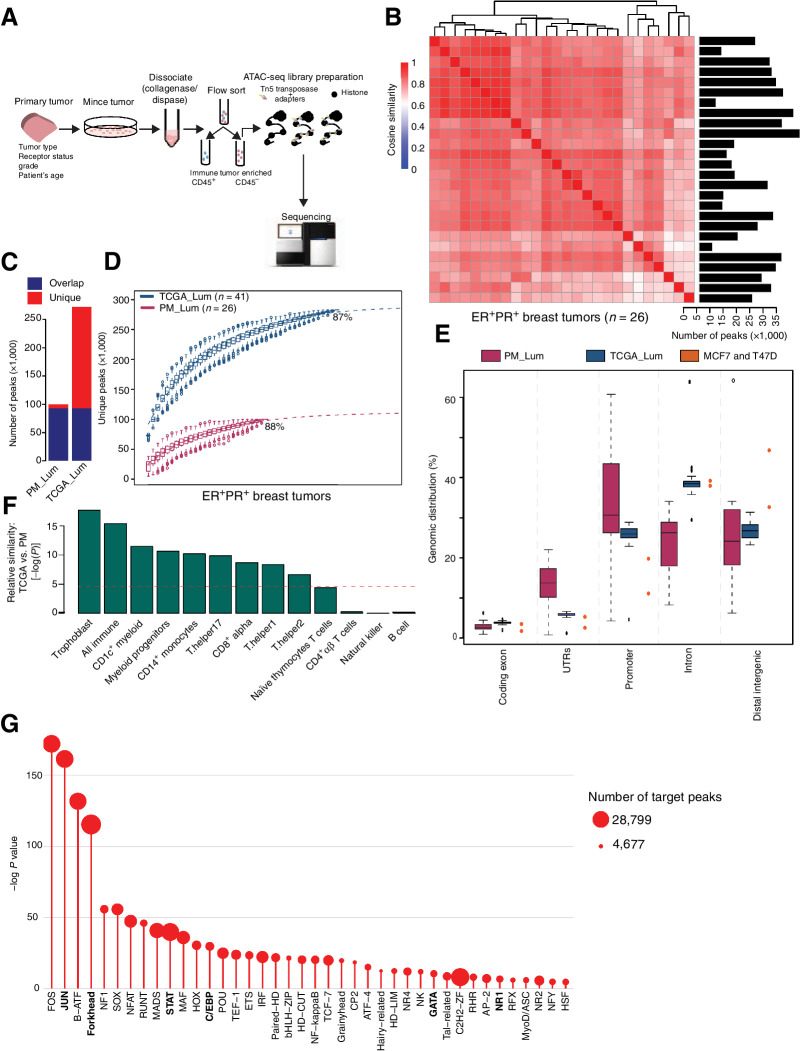
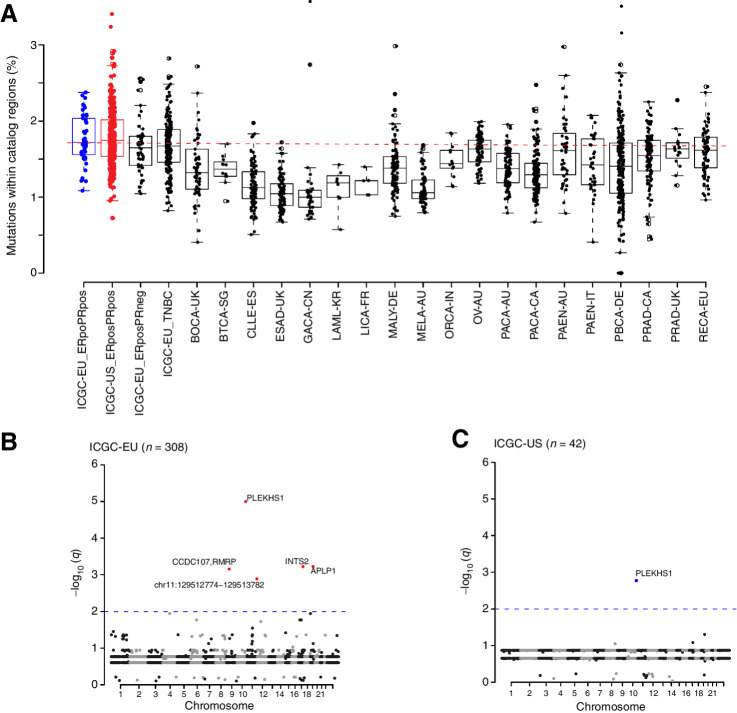
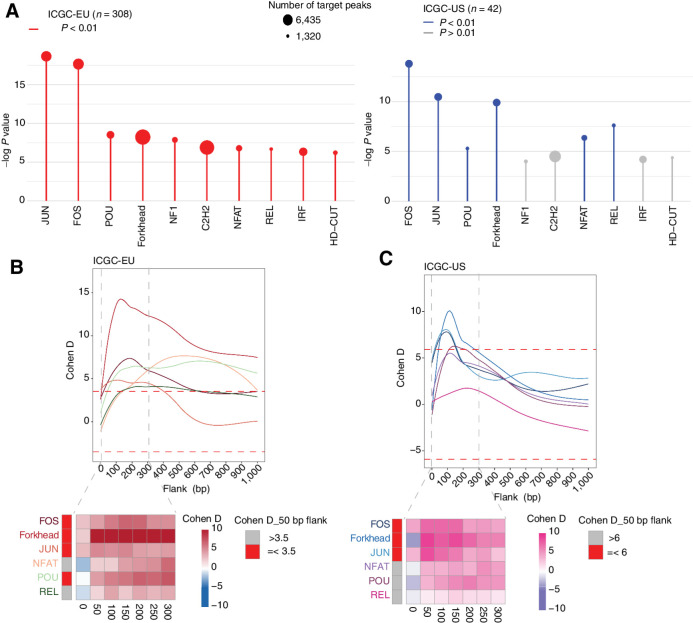
![Figure 4. High enrichment of mutations at cistromes of key transcription factors involved in ER+PR+ breast cancer. Heatmaps showing enrichment of mutations at ChIP-seq peak centers and flanking regions (0–1000 bp) using ICGC-EU WGS dataset (A), transcription factor–binding sets using ICGC-EU (B), and ICGC-US WGS datasets (C). Cohen D was calculated based on resampling and the value indicates significant enrichment [Enrichment > Median (Cohen D)]. Transcription factors showing a consensus in mutation enrichment are marked in bold.](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/9a7d/9398156/c241fe6245e0/102fig4.jpg)
References
-
- Aversa C, Rossi V, Geuna E, Martinello R, Milani A, Redana S, et al. Metastatic breast cancer subtypes and central nervous system metastases. Breast 2014;23:623–8. - PubMed
-
- Harvey JM, Clark GM, Osborne CK, Allred DC. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol 1999;17:1474–81. - PubMed
-
- Martínez-Jiménez F, Muiños F, Sentís I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, et al. A compendium of mutational cancer driver genes. Nat Rev Cancer 2020;20:555–72. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
