

TNF-α synergises with IFN-γ to induce caspase-8-JAK1/2-STAT1-dependent death of intestinal epithelial cells
- PMID: 34556638
- PMCID: PMC8459343
- DOI: 10.1038/s41419-021-04151-3
TNF-α synergises with IFN-γ to induce caspase-8-JAK1/2-STAT1-dependent death of intestinal epithelial cells
Abstract
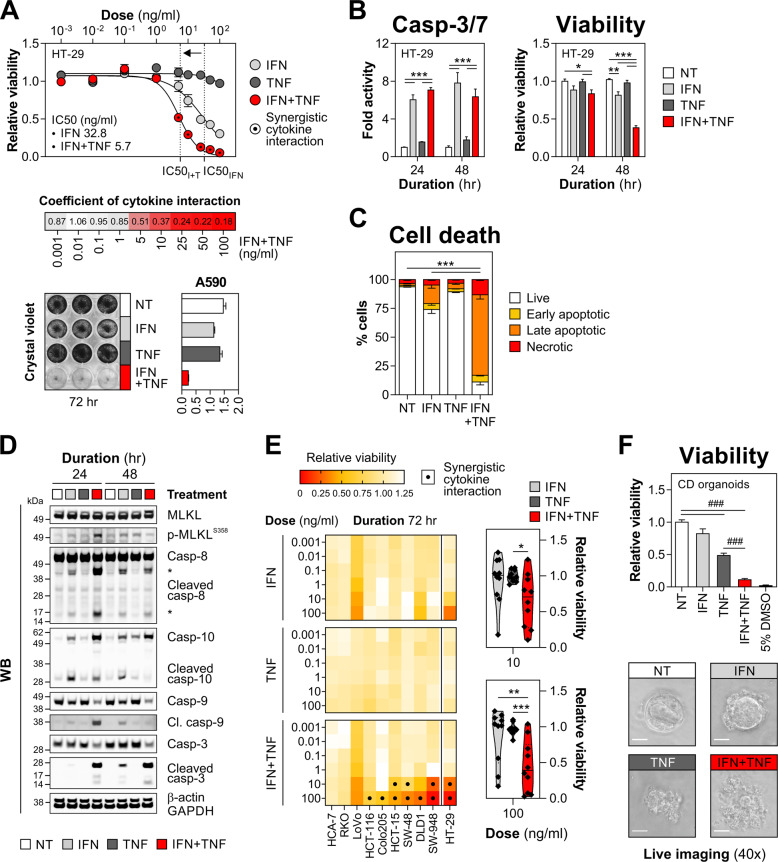
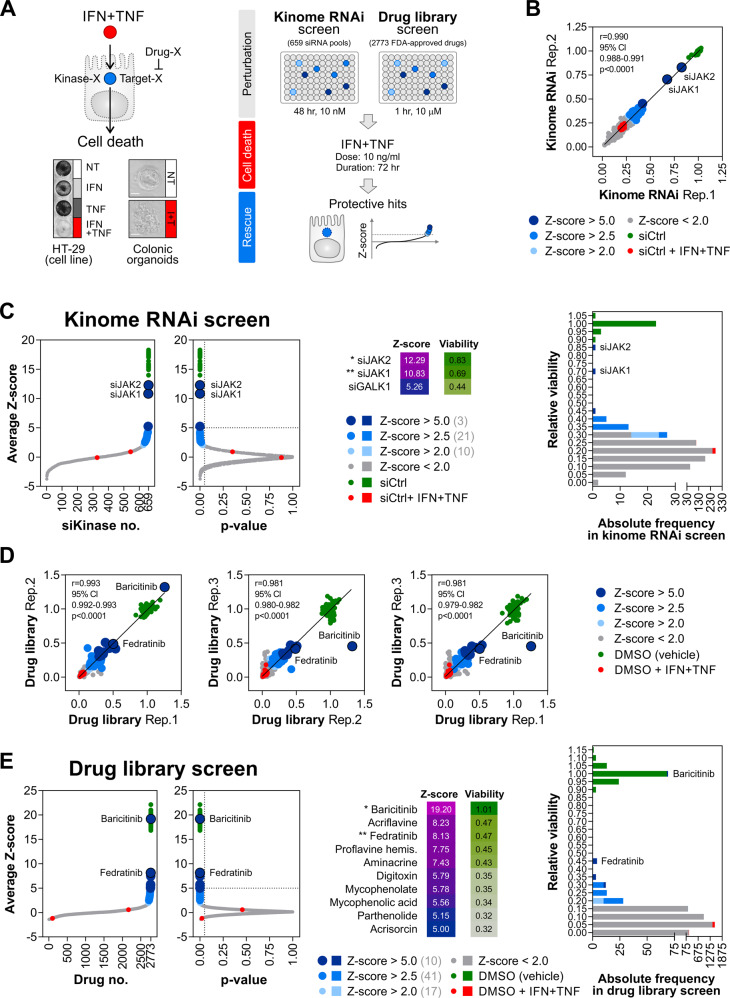
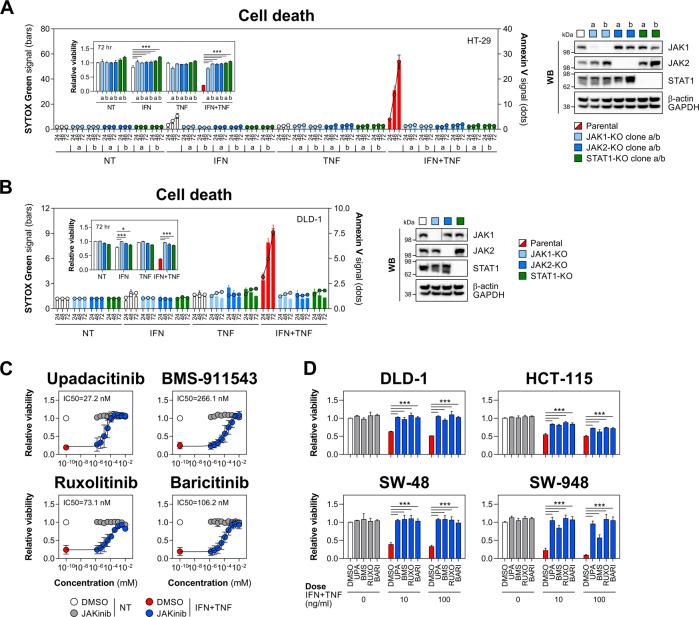
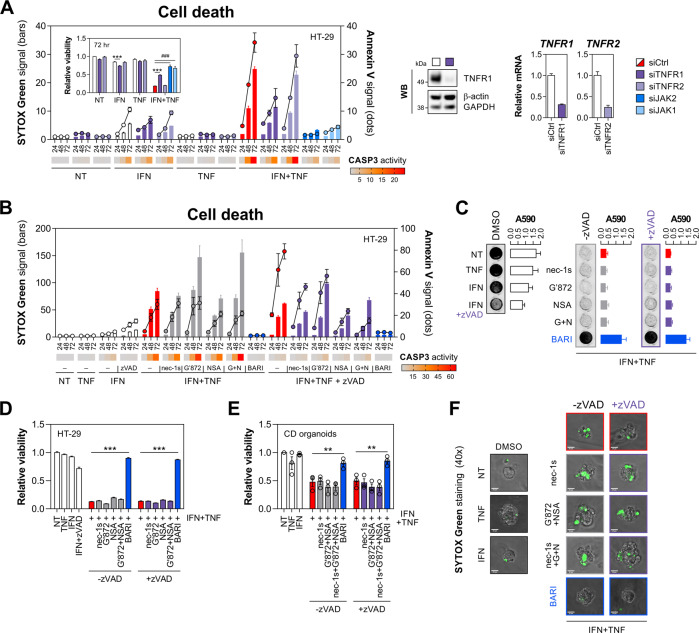
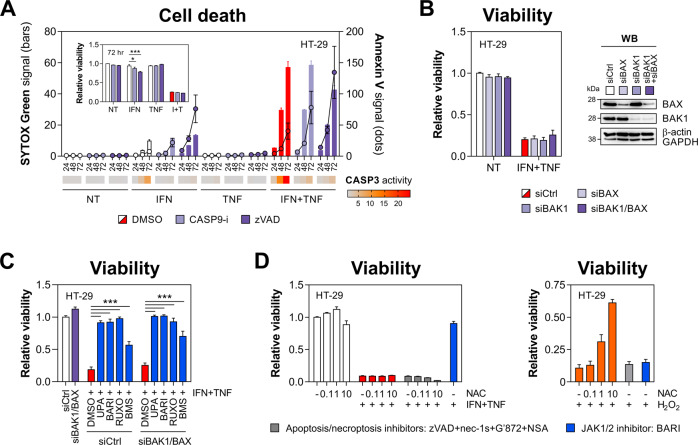
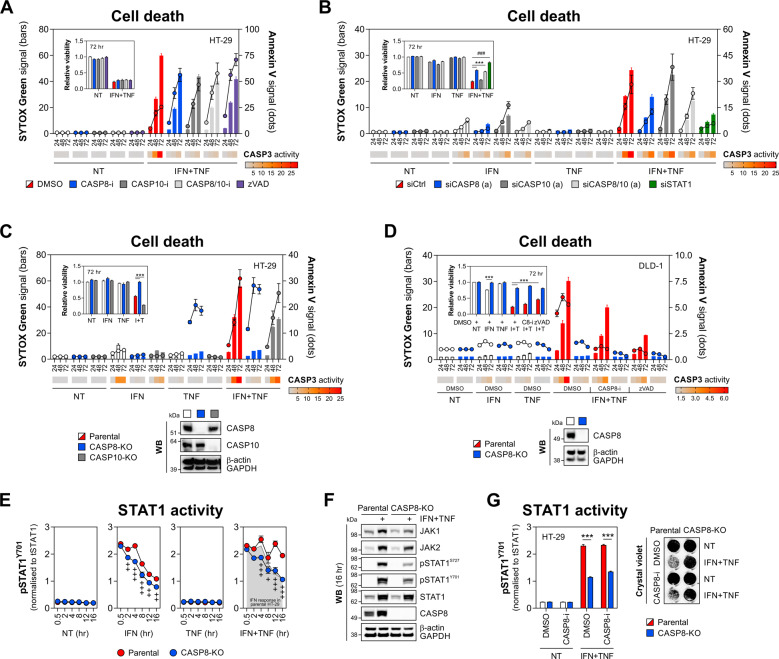
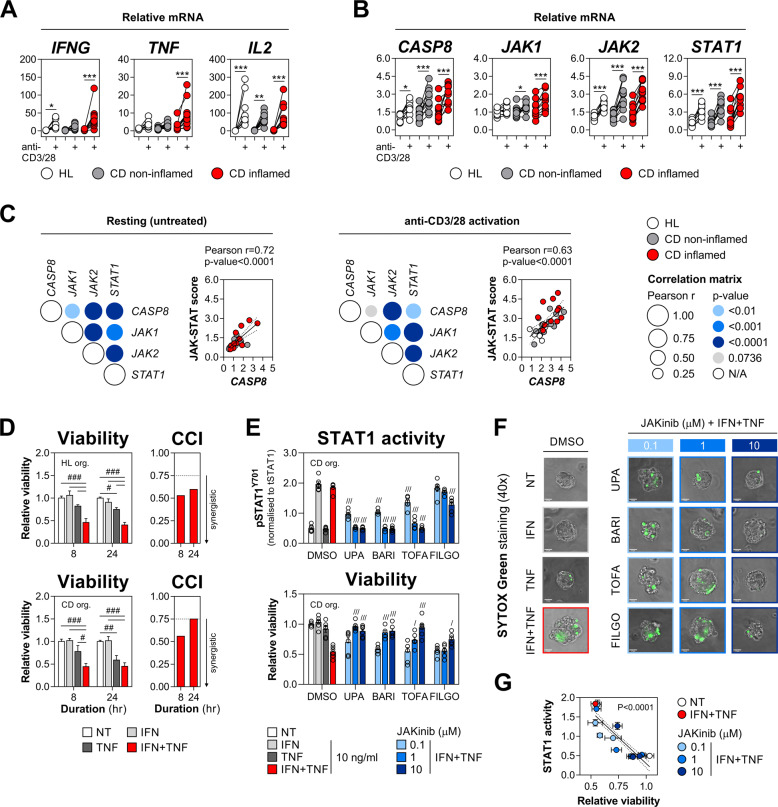
Rewiring of host cytokine networks is a key feature of inflammatory bowel diseases (IBD) such as Crohn's disease (CD). Th1-type cytokines-IFN-γ and TNF-α-occupy critical nodes within these networks and both are associated with disruption of gut epithelial barrier function. This may be due to their ability to synergistically trigger the death of intestinal epithelial cells (IECs) via largely unknown mechanisms. In this study, through unbiased kinome RNAi and drug repurposing screens we identified JAK1/2 kinases as the principal and nonredundant drivers of the synergistic killing of human IECs by IFN-γ/TNF-α. Sensitivity to IFN-γ/TNF-α-mediated synergistic IEC death was retained in primary patient-derived intestinal organoids. Dependence on JAK1/2 was confirmed using genetic loss-of-function studies and JAK inhibitors (JAKinibs). Despite the presence of biochemical features consistent with canonical TNFR1-mediated apoptosis and necroptosis, IFN-γ/TNF-α-induced IEC death was independent of RIPK1/3, ZBP1, MLKL or caspase activity. Instead, it involved sustained activation of JAK1/2-STAT1 signalling, which required a nonenzymatic scaffold function of caspase-8 (CASP8). Further modelling in gut mucosal biopsies revealed an intercorrelated induction of the lethal CASP8-JAK1/2-STAT1 module during ex vivo stimulation of T cells. Functional studies in CD-derived organoids using inhibitors of apoptosis, necroptosis and JAKinibs confirmed the causative role of JAK1/2-STAT1 in cytokine-induced death of primary IECs. Collectively, we demonstrate that TNF-α synergises with IFN-γ to kill IECs via the CASP8-JAK1/2-STAT1 module independently of canonical TNFR1 and cell death signalling. This non-canonical cell death pathway may underpin immunopathology driven by IFN-γ/TNF-α in diverse autoinflammatory diseases such as IBD, and its inhibition may contribute to the therapeutic efficacy of anti-TNFs and JAKinibs.
© 2021. The Author(s).
Conflict of interest statement
K.N. and F.S. are in receipt of research funding from AbbVie Inc. in the context of a research centre spoke award (SFI-14/SP/2710) to APC Microbiome Ireland. B.L.M. is an employee and shareholder of AbbVie Inc.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
