

Clinical and therapeutic significance of genetic variation in the GRIN gene family encoding NMDARs
- PMID: 34560056
- PMCID: PMC8525401
- DOI: 10.1016/j.neuropharm.2021.108805
Clinical and therapeutic significance of genetic variation in the GRIN gene family encoding NMDARs
Abstract
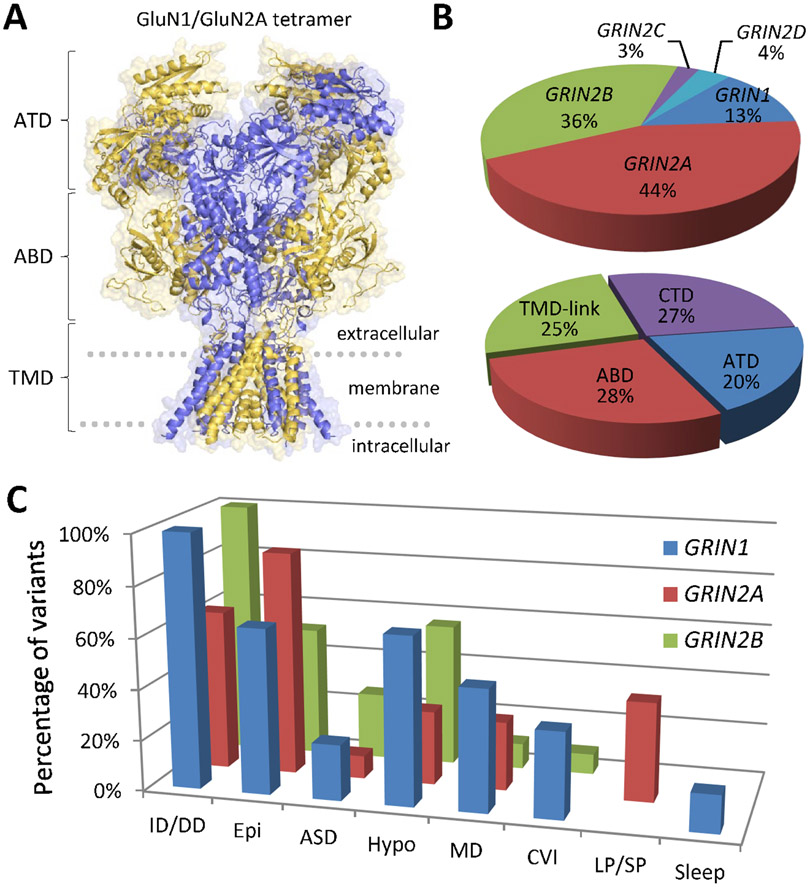
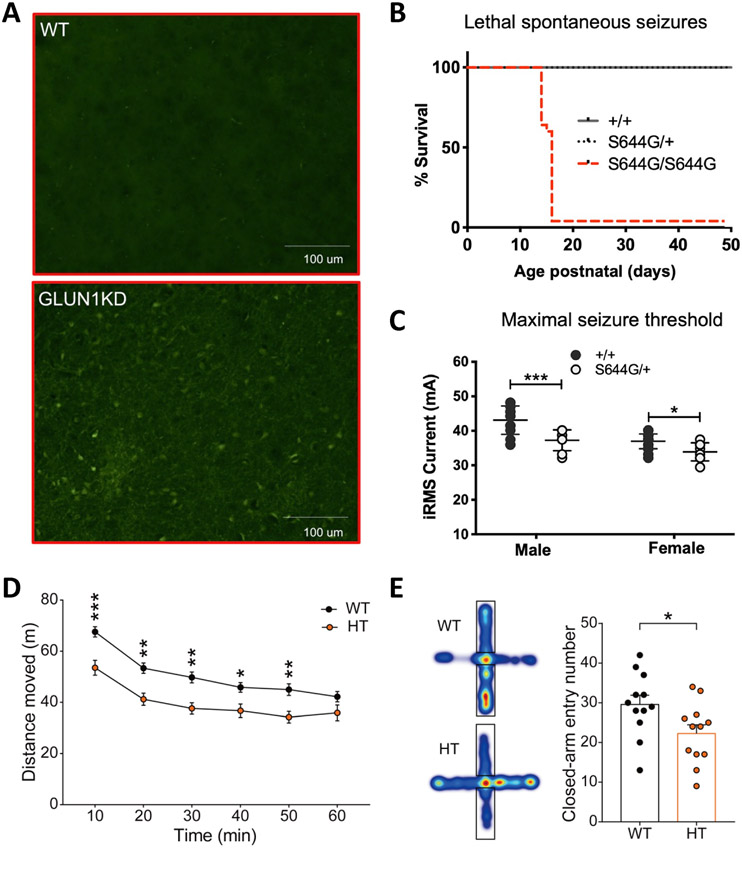
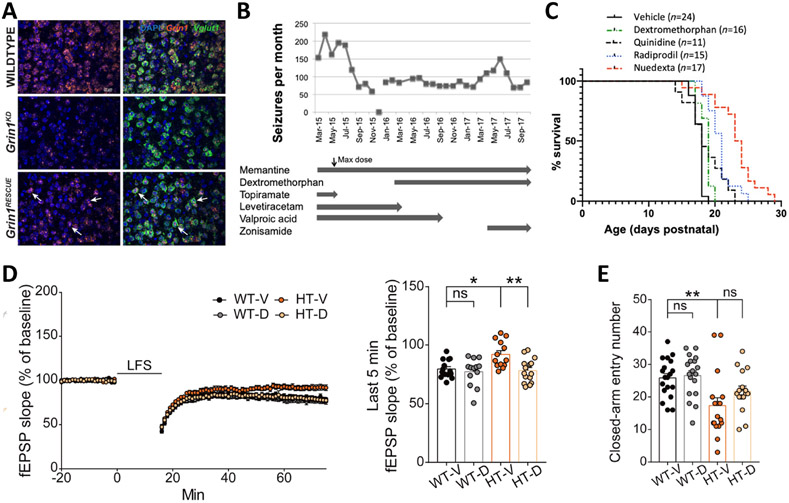
Considerable genetic variation of N-methyl-d-aspartate receptors (NMDARs) has recently become apparent, with many hundreds of de novo variants identified through widely available clinical genetic testing. Individuals with GRIN variants present with neurological conditions such as epilepsy, autism, intellectual disability (ID), movement disorders, schizophrenia and behavioral disorders. Determination of the functional consequence of genetic variation for NMDARs should lead to precision therapeutics. Furthermore, genetic animal models harboring human variants have the potential to reveal mechanisms that are shared among different neurological conditions, providing strategies that may allow treatment of individuals who are refractory to therapy. Preclinical studies in animal models and small open label trials in humans support this idea. However, additional functional data for variants and animal models corresponding to multiple individuals with the same genotype are needed to validate this approach and to lead to thoughtfully designed, randomized, placebo-controlled clinical trials, which could provide data in order to determine safety and efficacy of potential precision therapeutics.
Keywords: Epilepsy; GRIN; Intellectual disability; NMDARs.
Copyright © 2021 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Distinct roles of GRIN2A and GRIN2B variants in neurological conditions.F1000Res. 2019 Nov 20;8:F1000 Faculty Rev-1940. doi: 10.12688/f1000research.18949.1. eCollection 2019. F1000Res. 2019. PMID: 31807283 Free PMC article. Review.
-
Disease-Associated Variants in GRIN1, GRIN2A and GRIN2B genes: Insights into NMDA Receptor Structure, Function, and Pathophysiology.Physiol Res. 2024 May 31;73(Suppl 1):S413-S434. doi: 10.33549/physiolres.935346. Epub 2024 May 31. Physiol Res. 2024. PMID: 38836461 Free PMC article. Review.
-
Disease-associated GRIN protein truncating variants trigger NMDA receptor loss-of-function.Hum Mol Genet. 2021 Feb 25;29(24):3859-3871. doi: 10.1093/hmg/ddaa220. Hum Mol Genet. 2021. PMID: 33043365
-
Positive allosteric modulators that target NMDA receptors rectify loss-of-function GRIN variants associated with neurological and neuropsychiatric disorders.Neuropharmacology. 2020 Oct 15;177:108247. doi: 10.1016/j.neuropharm.2020.108247. Epub 2020 Jul 24. Neuropharmacology. 2020. PMID: 32712275 Free PMC article.
-
De novo GRIN variants in NMDA receptor M2 channel pore-forming loop are associated with neurological diseases.Hum Mutat. 2019 Dec;40(12):2393-2413. doi: 10.1002/humu.23895. Epub 2019 Sep 10. Hum Mutat. 2019. PMID: 31429998 Free PMC article.
Cited by
-
NMDA Receptor C-Terminal Domain Signalling in Development, Maturity, and Disease.Int J Mol Sci. 2022 Sep 27;23(19):11392. doi: 10.3390/ijms231911392. Int J Mol Sci. 2022. PMID: 36232696 Free PMC article. Review.
-
Comprehensive Transcriptomic Investigation of Rett Syndrome Reveals Increasing Complexity Trends from Induced Pluripotent Stem Cells to Neurons with Implications for Enriched Pathways.ACS Omega. 2023 Nov 8;8(46):44148-44162. doi: 10.1021/acsomega.3c06448. eCollection 2023 Nov 21. ACS Omega. 2023. PMID: 38027357 Free PMC article.
-
Disruption of the Ubiquitin-Proteasome System and Elevated Endoplasmic Reticulum Stress in Epilepsy.Biomedicines. 2022 Mar 11;10(3):647. doi: 10.3390/biomedicines10030647. Biomedicines. 2022. PMID: 35327449 Free PMC article. Review.
-
Disease-associated nonsense and frame-shift variants resulting in the truncation of the GluN2A or GluN2B C-terminal domain decrease NMDAR surface expression and reduce potentiating effects of neurosteroids.Cell Mol Life Sci. 2024 Jan 12;81(1):36. doi: 10.1007/s00018-023-05062-6. Cell Mol Life Sci. 2024. PMID: 38214768 Free PMC article.
-
Spermidine Treatment Improves GRIN2B Loss-Of-Function, A Primary Disorder of Glutamatergic Neurotransmission.J Inherit Metab Dis. 2025 Mar;48(2):e70015. doi: 10.1002/jimd.70015. J Inherit Metab Dis. 2025. PMID: 40024627 Free PMC article.
References
-
- Paciorkowski AR, Seltzer LE, and Neul JL, Developmental Encephalopathies, in Swaiman's Pediatric Neurology, Swaiman KF, et al., Editors. 2018, Mosby: Philadelphia. p. 242–248.
-
- Institute of Medicine (U.S.). Committee to Evaluate the Supplemental Security Income Disability Program for Children with Mental Disorders, et al., Mental disorders and disabilities among low-income children. 2015, National Academies Press: Washington, D.C. p. 169–178. - PubMed
-
- McTague A, et al., The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol, 2016. 15(3): p. 304–16. - PubMed
