

Large-scale data analysis for robotic yeast one-hybrid platforms and multi-disciplinary studies using GateMultiplex
- PMID: 34560855
- PMCID: PMC8461970
- DOI: 10.1186/s12915-021-01140-y
Large-scale data analysis for robotic yeast one-hybrid platforms and multi-disciplinary studies using GateMultiplex
Abstract
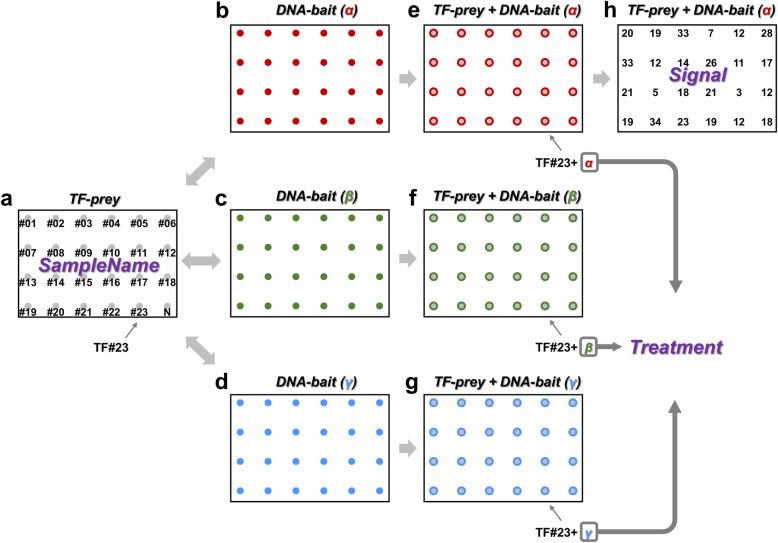
Background: Yeast one-hybrid (Y1H) is a common technique for identifying DNA-protein interactions, and robotic platforms have been developed for high-throughput analyses to unravel the gene regulatory networks in many organisms. Use of these high-throughput techniques has led to the generation of increasingly large datasets, and several software packages have been developed to analyze such data. We previously established the currently most efficient Y1H system, meiosis-directed Y1H; however, the available software tools were not designed for processing the additional parameters suggested by meiosis-directed Y1H to avoid false positives and required programming skills for operation.
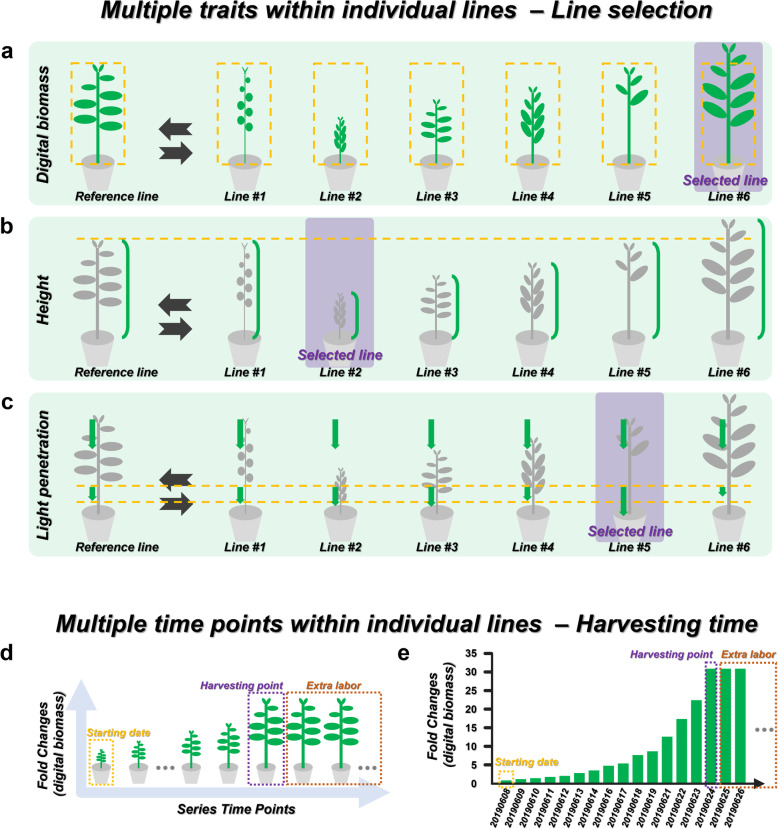
Results: We developed a new tool named GateMultiplex with high computing performance using C++. GateMultiplex incorporated a graphical user interface (GUI), which allows the operation without any programming skills. Flexible parameter options were designed for multiple experimental purposes to enable the application of GateMultiplex even beyond Y1H platforms. We further demonstrated the data analysis from other three fields using GateMultiplex, the identification of lead compounds in preclinical cancer drug discovery, the crop line selection in precision agriculture, and the ocean pollution detection from deep-sea fishery.
Conclusions: The user-friendly GUI, fast C++ computing speed, flexible parameter setting, and applicability of GateMultiplex facilitate the feasibility of large-scale data analysis in life science fields.
Keywords: C++; Deep-sea fishery; Precision agriculture; Preclinical drug discovery; Yeast one-hybrid.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures











References
-
- Wang Z, Mao Y, Guo Y, Gao J, Liu X, Li S, Lin YCJ, Chen H, Wang JP, Chiang VL, Li W. MYB transcription factor161 mediates feedback regulation of secondary wall-associated NAC-Domain1 family genes for wood formation. Plant Physiol. 2020;184(3):1389–1406. doi: 10.1104/pp.20.01033. - DOI - PMC - PubMed
-
- Yeh CS, Wang Z, Miao F, Ma H, Kao CT, Hsu TS, Yu JH, Hung ET, Lin CC, Kuan CY, Tsai NC, Zhou C, Qu GZ, Jiang J, Liu G, Wang JP, Li W, Chiang VL, Chang TH, Lin YCJ. A novel synthetic-genetic-array-based yeast one-hybrid system for high discovery rate and short processing time. Genome Res. 2019;29(8):1343–1351. doi: 10.1101/gr.245951.118. - DOI - PMC - PubMed
-
- Lin YC, Li W, Sun YH, Kumari S, Wei H, Li Q, Tunlaya-Anukit S, Sederoff RR, Chiang VL. SND1 transcription factor-directed quantitative functional hierarchical genetic regulatory network in wood formation in Populus trichocarpa. Plant Cell. 2013;25(11):4324–4341. doi: 10.1105/tpc.113.117697. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
