

What's Wrong in a Jump? Prediction and Validation of Splice Site Variants
- PMID: 34564308
- PMCID: PMC8482176
- DOI: 10.3390/mps4030062
What's Wrong in a Jump? Prediction and Validation of Splice Site Variants
Abstract
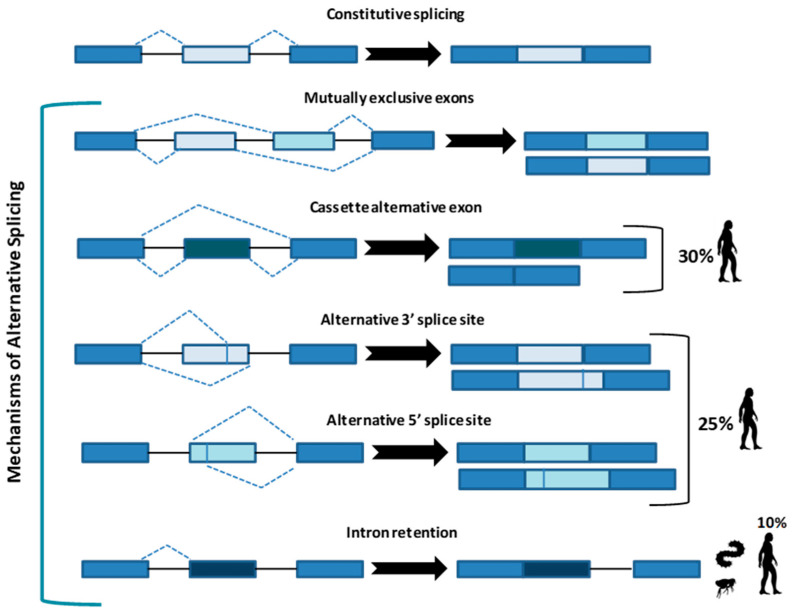
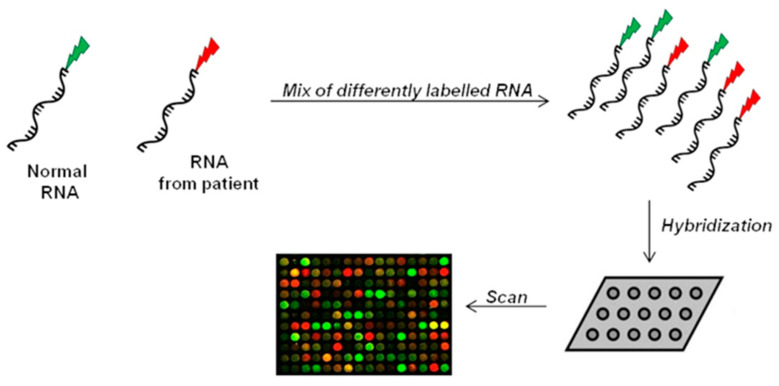
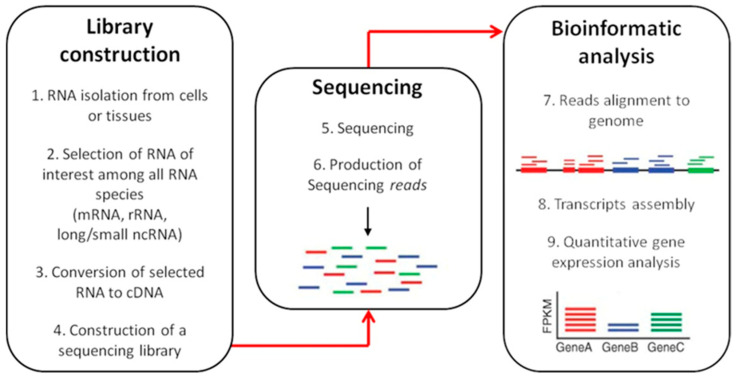
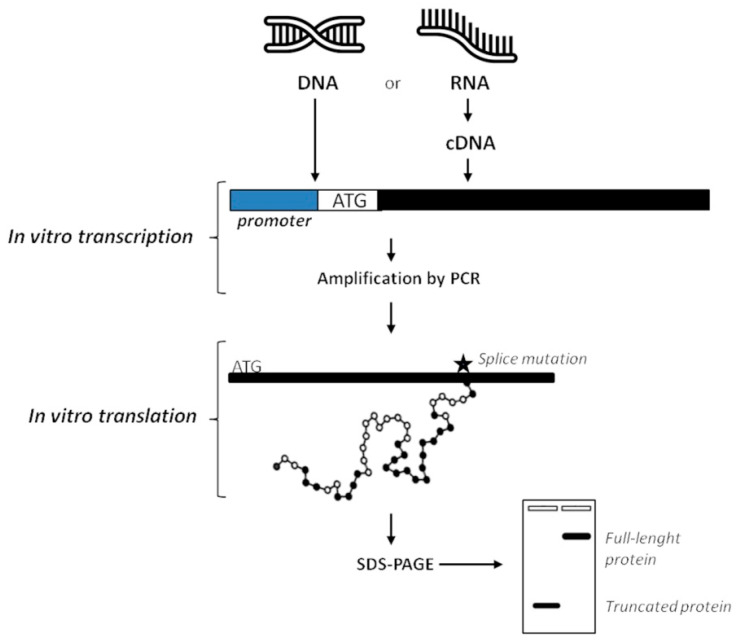
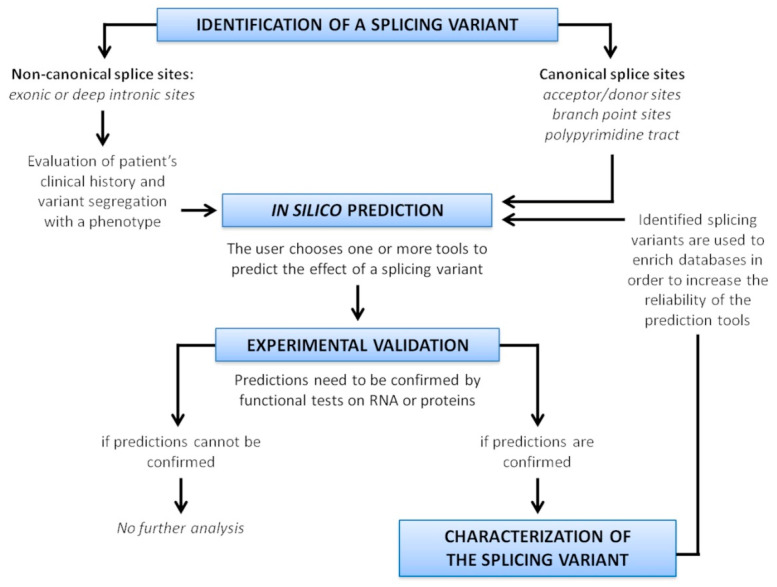
Alternative splicing (AS) is a crucial process to enhance gene expression driving organism development. Interestingly, more than 95% of human genes undergo AS, producing multiple protein isoforms from the same transcript. Any alteration (e.g., nucleotide substitutions, insertions, and deletions) involving consensus splicing regulatory sequences in a specific gene may result in the production of aberrant and not properly working proteins. In this review, we introduce the key steps of splicing mechanism and describe all different types of genomic variants affecting this process (splicing variants in acceptor/donor sites or branch point or polypyrimidine tract, exonic, and deep intronic changes). Then, we provide an updated approach to improve splice variants detection. First, we review the main computational tools, including the recent Machine Learning-based algorithms, for the prediction of splice site variants, in order to characterize how a genomic variant interferes with splicing process. Next, we report the experimental methods to validate the predictive analyses are defined, distinguishing between methods testing RNA (transcriptomics analysis) or proteins (proteomics experiments). For both prediction and validation steps, benefits and weaknesses of each tool/procedure are accurately reported, as well as suggestions on which approaches are more suitable in diagnostic rather than in clinical research.
Keywords: alternative splicing; experimental validation; machine learning; prediction tools; splice variant; splicing sites; variant classification.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–423. doi: 10.1038/gim.2015.30. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
