

Can the Mitochondrial Metabolic Theory Explain Better the Origin and Management of Cancer than Can the Somatic Mutation Theory?
- PMID: 34564387
- PMCID: PMC8467939
- DOI: 10.3390/metabo11090572
Can the Mitochondrial Metabolic Theory Explain Better the Origin and Management of Cancer than Can the Somatic Mutation Theory?
Abstract
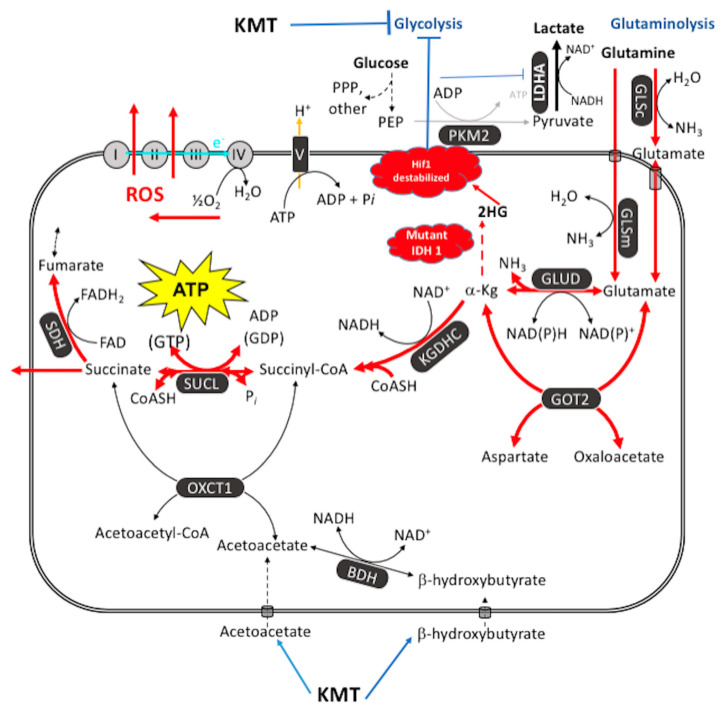
A theory that can best explain the facts of a phenomenon is more likely to advance knowledge than a theory that is less able to explain the facts. Cancer is generally considered a genetic disease based on the somatic mutation theory (SMT) where mutations in proto-oncogenes and tumor suppressor genes cause dysregulated cell growth. Evidence is reviewed showing that the mitochondrial metabolic theory (MMT) can better account for the hallmarks of cancer than can the SMT. Proliferating cancer cells cannot survive or grow without carbons and nitrogen for the synthesis of metabolites and ATP (Adenosine Triphosphate). Glucose carbons are essential for metabolite synthesis through the glycolysis and pentose phosphate pathways while glutamine nitrogen and carbons are essential for the synthesis of nitrogen-containing metabolites and ATP through the glutaminolysis pathway. Glutamine-dependent mitochondrial substrate level phosphorylation becomes essential for ATP synthesis in cancer cells that over-express the glycolytic pyruvate kinase M2 isoform (PKM2), that have deficient OxPhos, and that can grow in either hypoxia (0.1% oxygen) or in cyanide. The simultaneous targeting of glucose and glutamine, while elevating levels of non-fermentable ketone bodies, offers a simple and parsimonious therapeutic strategy for managing most cancers.
Keywords: IDH1; chimpanzees; evolution; fermentation; glutaminolysis; glycolysis; ketogenic metabolic therapy; metastasis; mitochondrial substrate level phosphorylation; mutations; oncogenes; respiration.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
LinkOut - more resources
Full Text Sources
Miscellaneous
