

Proteomes Are of Proteoforms: Embracing the Complexity
- PMID: 34564541
- PMCID: PMC8482110
- DOI: 10.3390/proteomes9030038
Proteomes Are of Proteoforms: Embracing the Complexity
Abstract
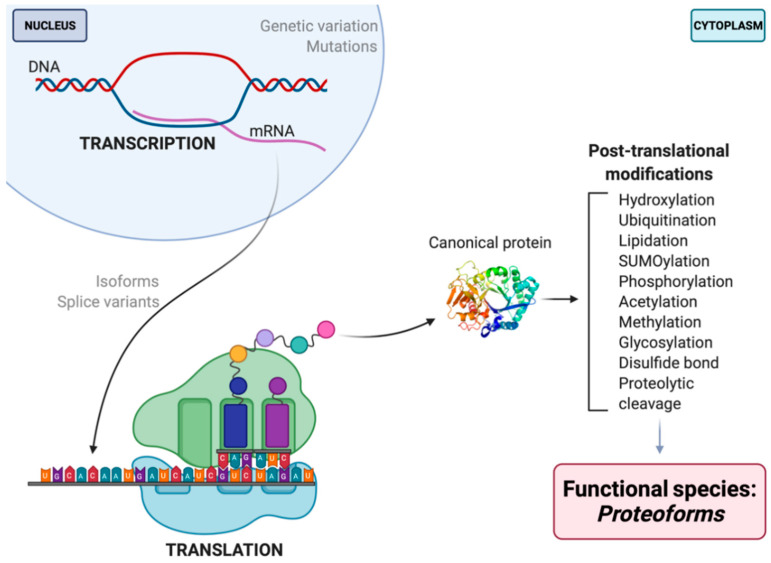
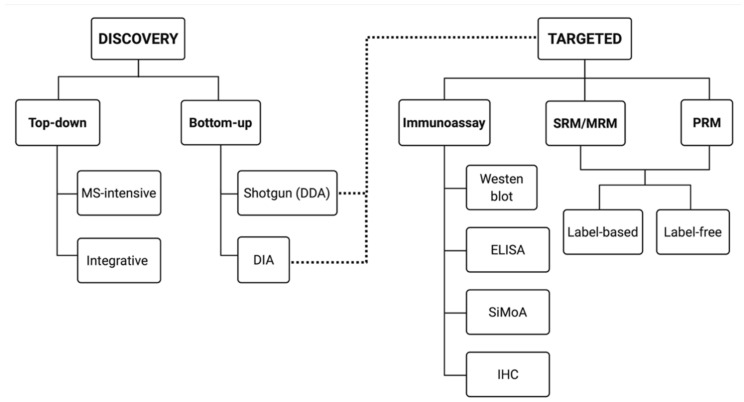
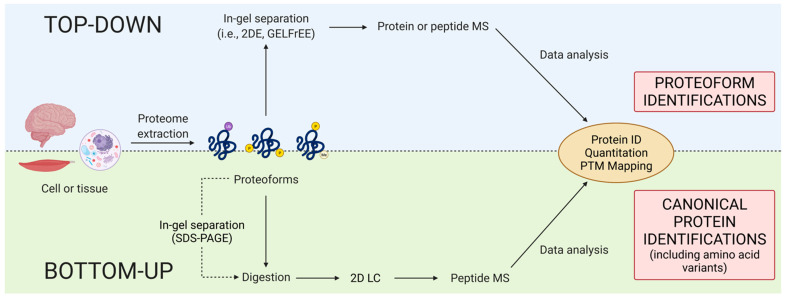
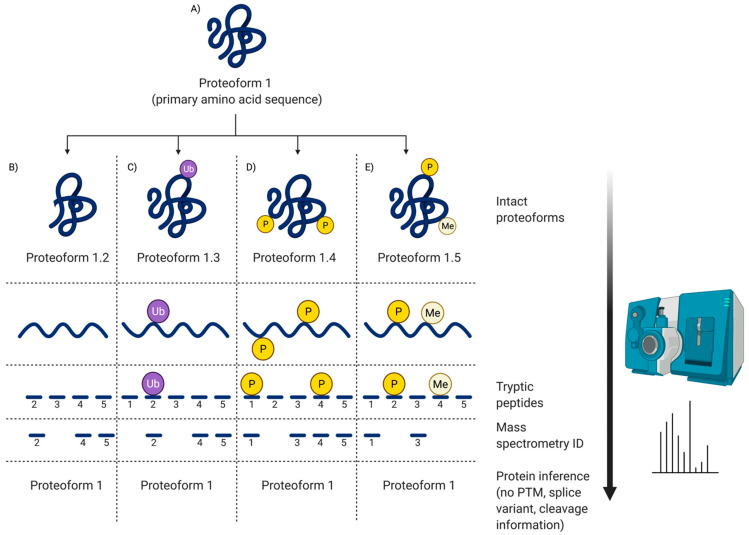
Proteomes are complex-much more so than genomes or transcriptomes. Thus, simplifying their analysis does not simplify the issue. Proteomes are of proteoforms, not canonical proteins. While having a catalogue of amino acid sequences provides invaluable information, this is the Proteome-lite. To dissect biological mechanisms and identify critical biomarkers/drug targets, we must assess the myriad of proteoforms that arise at any point before, after, and between translation and transcription (e.g., isoforms, splice variants, and post-translational modifications [PTM]), as well as newly defined species. There are numerous analytical methods currently used to address proteome depth and here we critically evaluate these in terms of the current 'state-of-the-field'. We thus discuss both pros and cons of available approaches and where improvements or refinements are needed to quantitatively characterize proteomes. To enable a next-generation approach, we suggest that advances lie in transdisciplinarity via integration of current proteomic methods to yield a unified discipline that capitalizes on the strongest qualities of each. Such a necessary (if not revolutionary) shift cannot be accomplished by a continued primary focus on proteo-genomics/-transcriptomics. We must embrace the complexity. Yes, these are the hard questions, and this will not be easy…but where is the fun in easy?
Keywords: Western blotting; bottom-up; immunoassay; mass spectrometry; proteomics; top-down; two-dimensional gel electrophoresis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

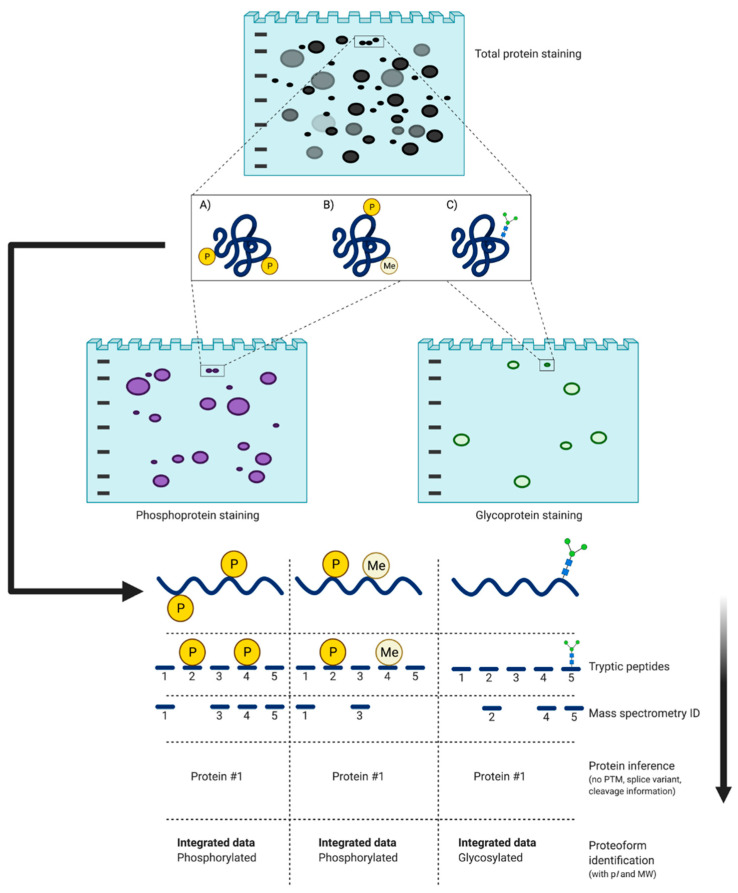
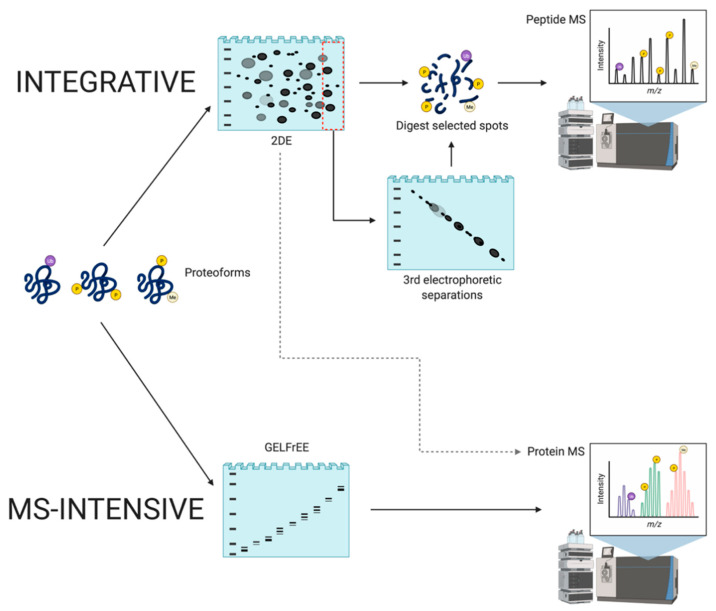
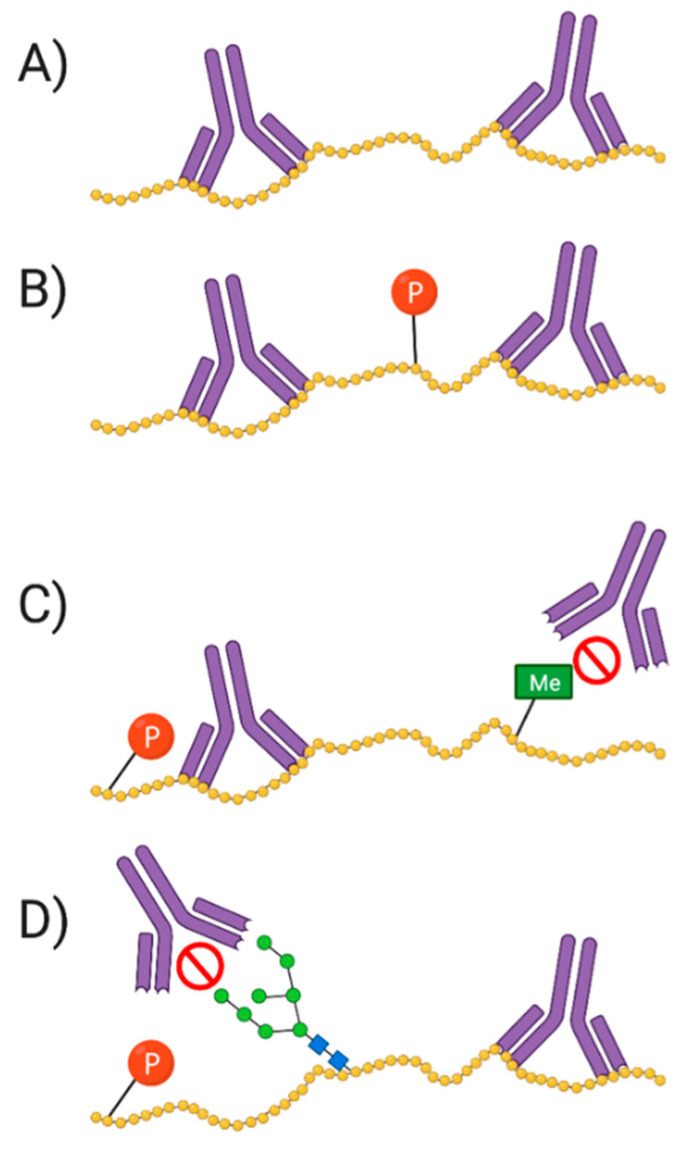
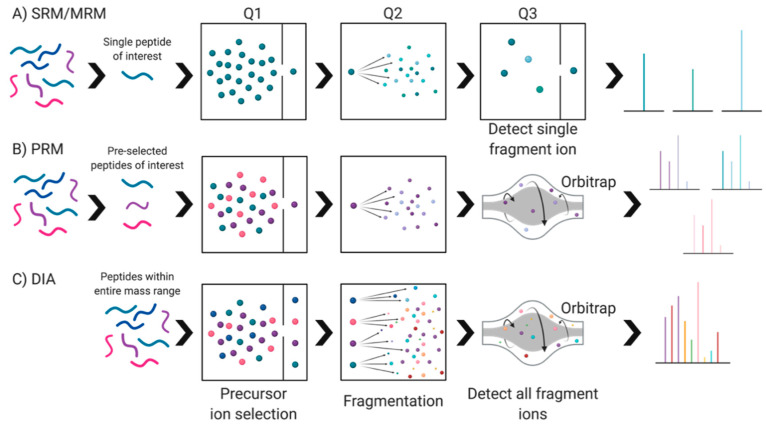
References
-
- Wilkins M.R., Sanchez J.-C., Gooley A.A., Appel R.D., Humphery-Smith I., Hochstrasser D.F., Williams K.L. Progress with proteome projects: Why all proteins expressed by a genome should be identified and how to do it. Biotechnol. Genet. Eng. Rev. 1996;13:19–50. doi: 10.1080/02648725.1996.10647923. - DOI - PubMed
-
- Duncan M.W., Yergey A.L., Gale P.J., Kate Y. Quantifying proteins by mass spectrometry. LC-GC N. Am. 2014;32:726–735.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
