

Carnitine is a pharmacological allosteric chaperone of the human lysosomal α-glucosidase
- PMID: 34565280
- PMCID: PMC8477953
- DOI: 10.1080/14756366.2021.1975694
Carnitine is a pharmacological allosteric chaperone of the human lysosomal α-glucosidase
Abstract
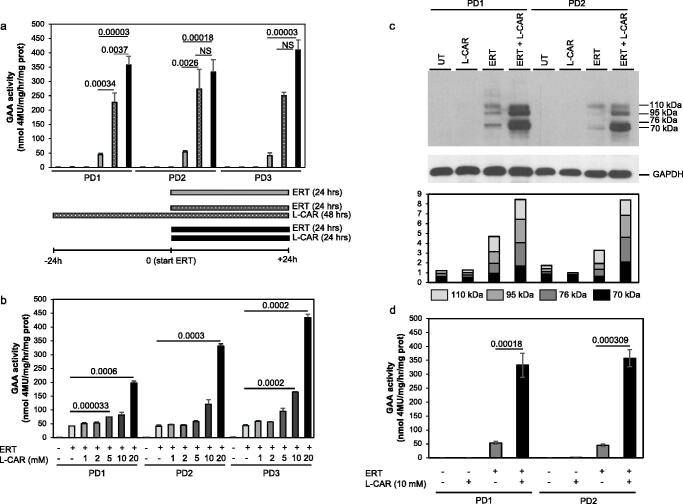
Pompe disease is an inherited metabolic disorder due to the deficiency of the lysosomal acid α-glucosidase (GAA). The only approved treatment is enzyme replacement therapy with the recombinant enzyme (rhGAA). Further approaches like pharmacological chaperone therapy, based on the stabilising effect induced by small molecules on the target enzyme, could be a promising strategy. However, most known chaperones could be limited by their potential inhibitory effects on patient's enzymes. Here we report on the discovery of novel chaperones for rhGAA, L- and D-carnitine, and the related compound acetyl-D-carnitine. These drugs stabilise the enzyme at pH and temperature without inhibiting the activity and acted synergistically with active-site directed pharmacological chaperones. Remarkably, they enhanced by 4-fold the acid α-glucosidase activity in fibroblasts from three Pompe patients with added rhGAA. This synergistic effect of L-carnitine and rhGAA has the potential to be translated into improved therapeutic efficacy of ERT in Pompe disease.
Keywords: carbohydrate active enzymes; glycogen storage disease type 2; lysosomal disease; orphan drugs; α-Glucosidase.
Conflict of interest statement
No potential conflict of interest was reported by the authors.
Figures







References
-
- van der Ploeg AT, Reuser AJ.. Pompe's disease. Lancet 2008;372:1342–53. - PubMed
-
- Parenti G, Andria G.. Pompe disease: from new views on pathophysiology to innovative therapeutic strategies. Curr Pharma Biotechnol 2011;12:902–15. - PubMed
-
- Parenti G, Moracci M, Fecarotta S, Andria G.. Pharmacological chaperone therapy for lysosomal storage diseases. Future Med Chem 2014;6:1031–45. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous