

pH-Dependent Conformations of an Antimicrobial Spider Venom Peptide, Cupiennin 1a, from Unbiased HREMD Simulations
- PMID: 34568695
- PMCID: PMC8459419
- DOI: 10.1021/acsomega.1c03729
pH-Dependent Conformations of an Antimicrobial Spider Venom Peptide, Cupiennin 1a, from Unbiased HREMD Simulations
Abstract
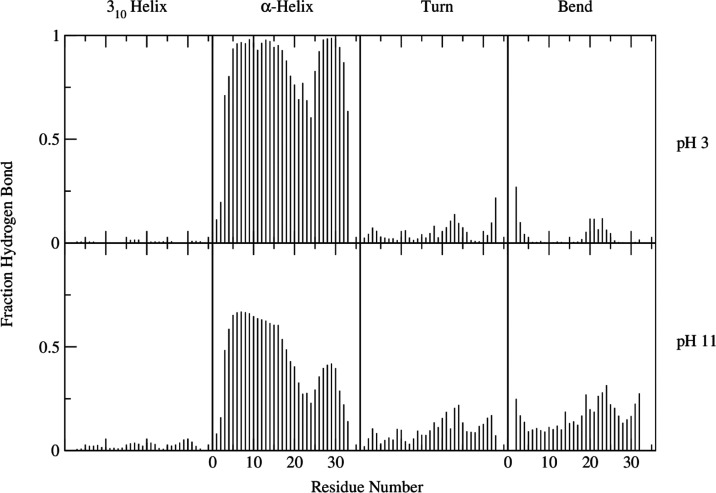
Cupiennin 1a is an antimicrobial peptide found in the venom of the spider Cupiennius salei. A highly cationic peptide, its cell lysis activity has been found to vary between neutral and charged membranes. In this study, Hamiltonian replica-exchange molecular dynamics (HREMD) was used to determine the conformational ensemble of the peptide in both charged (pH 3) and neutral (pH 11) states. The obtained free energy landscapes demonstrated the conformational diversity of the neutral peptide. At high pH, the peptide was found to adopt helix-hinge-helix and disordered structures. At pH 3, the peptide is structured with a high propensity toward α-helices. The presence of these α-helices seems to assist the peptide in recognizing membrane surfaces. These results highlight the importance of the charged residues in the stabilization of the peptide structure and the subsequent effects of pH on the peptide's conformational diversity and membrane activity. These findings may provide insights into the antimicrobial activity of Cupiennin 1a and other amphipathic linear peptides toward different cell membranes.
© 2021 The Authors. Published by American Chemical Society.
Conflict of interest statement
The authors declare no competing financial interest.
Figures






References
-
- Ferreira F. R. B.; da Silva P. M.; Soares T.; Machado L. G.; de Araújo L. C. C.; da Silva T. G.; de Mello G. S. V.; da Rocha Pitta M. G.; de Melo Rego M. J. B.; Pontual E. V.; Zingali R. B.; Napoleão T. H.; Paiva P. M. G. Evaluation of antimicrobial, cytotoxic, and hemolytic activities from venom of the spider Lasiodora sp. Toxicon 2016, 122, 119–126. 10.1016/j.toxicon.2016.09.019. - DOI - PubMed
-
- Mourão C. B. F.; Heghinian M. D.; Barbosa E. A.; Marí F.; Bloch C.; Restano-Cassulini R.; Possani L. D.; Schwartz E. F. Characterization of a Novel Peptide Toxin from Acanthoscurria paulensis Spider Venom: A Distinct Cysteine Assignment to the HWTX-II Family. Biochemistry 2013, 52, 2440–2452. 10.1021/bi4000035. - DOI - PubMed
-
- Undheim E.; Sunagar K.; Herzig V.; Kely L.; Low D.; Jackson T.; Jones A.; Kurniawan N.; King G.; Ali S.; Antunes A.; Ruder T.; Fry B. A Proteomics and Transcriptomics Investigation of the Venom from the Barychelid Spider Trittame loki (Brush-Foot Trapdoor). Toxins 2013, 5, 2488–2503. 10.3390/toxins5122488. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
