

Ferroptosis Meets Cell-Cell Contacts
- PMID: 34572111
- PMCID: PMC8471828
- DOI: 10.3390/cells10092462
Ferroptosis Meets Cell-Cell Contacts
Abstract
Ferroptosis is a regulated form of cell death characterized by iron dependency and increased lipid peroxidation. Initially assumed to be selectively induced in tumour cells, there is increasing evidence that ferroptosis plays an important role in pathophysiology and numerous cell types and tissues. Deregulated ferroptosis has been linked to human diseases, such as neurodegenerative diseases, cardiovascular disorders, and cancer. Along these lines, ferroptosis is a promising pathway to overcoming therapy resistance of cancer cells. It is therefore of utmost importance to understand the cellular signalling pathways and the molecular mechanisms underlying ferroptosis regulation, including context-specific effects mediated by the neighbouring cells through cell-cell contacts. Here, we give an overview on the molecular events and machinery linked to ferroptosis induction and commitment. We further summarize and discuss current knowledge about the role of cell-cell contacts, which differ in ferroptosis regulation between normal somatic cells and cancer cells. We present emerging concepts on the underlying mechanisms, address open questions, and discuss the possible impact of cell-cell contacts on exploiting ferroptosis in cancer therapy.
Keywords: cancer therapy; cell–cell contacts; epithelial–mesenchymal transition; ferroptosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
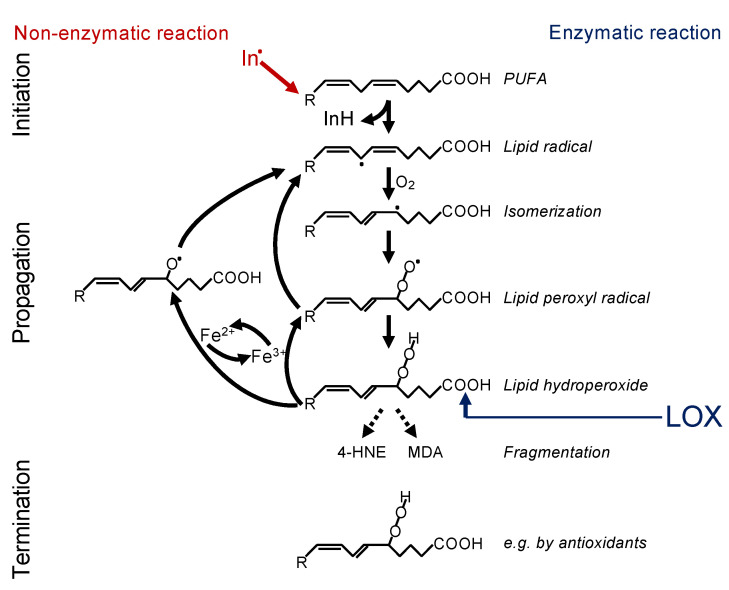
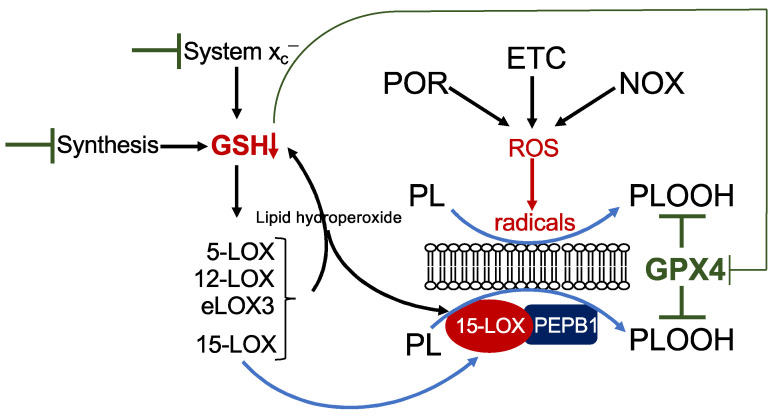
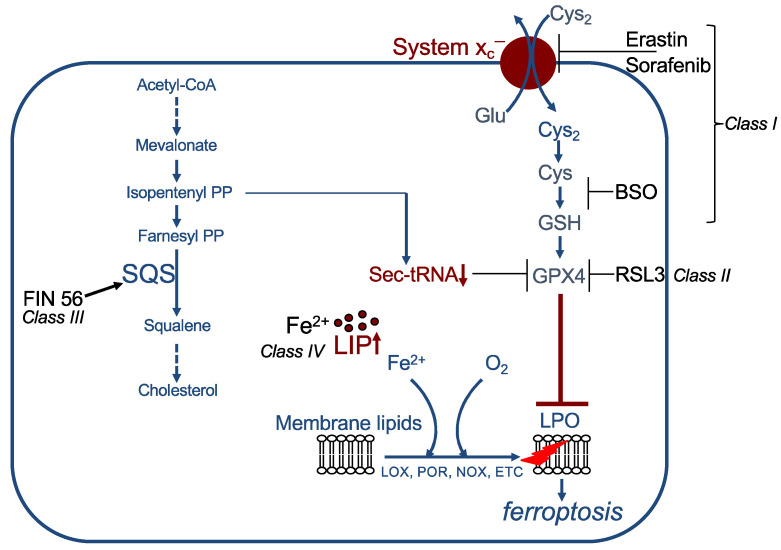
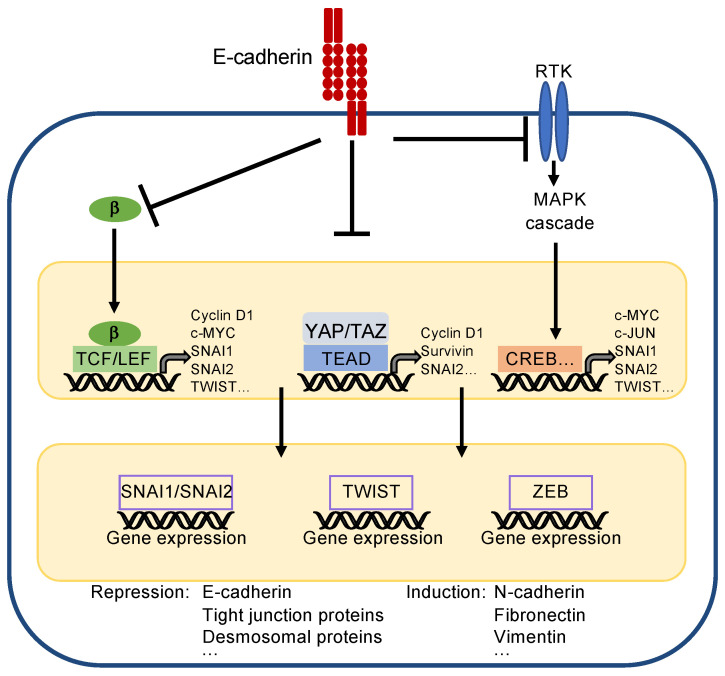
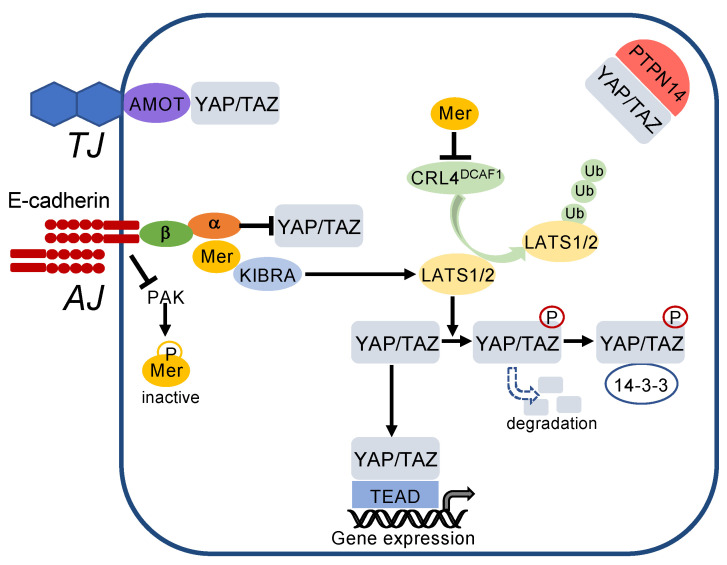
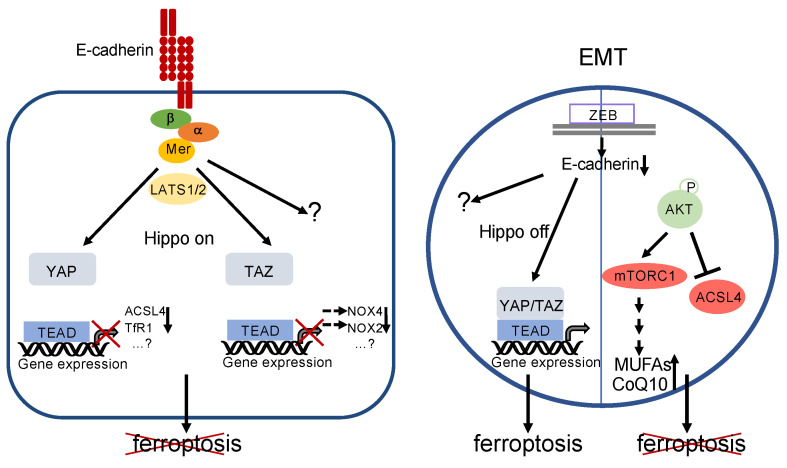
References
-
- Galluzzi L., Vitale I., Aaronson S.A., Abrams J.M., Adam D., Agostinis P., Alnemri E.S., Altucci L., Amelio I., Andrews D.W., et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
