

The Role of Smoothened-Dependent and -Independent Hedgehog Signaling Pathway in Tumorigenesis
- PMID: 34572373
- PMCID: PMC8466551
- DOI: 10.3390/biomedicines9091188
The Role of Smoothened-Dependent and -Independent Hedgehog Signaling Pathway in Tumorigenesis
Abstract
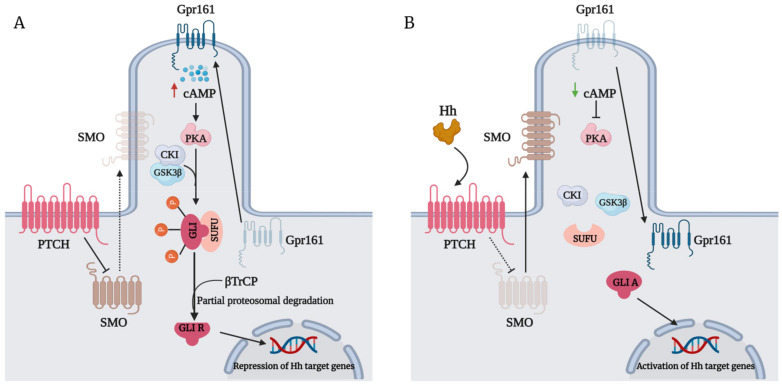
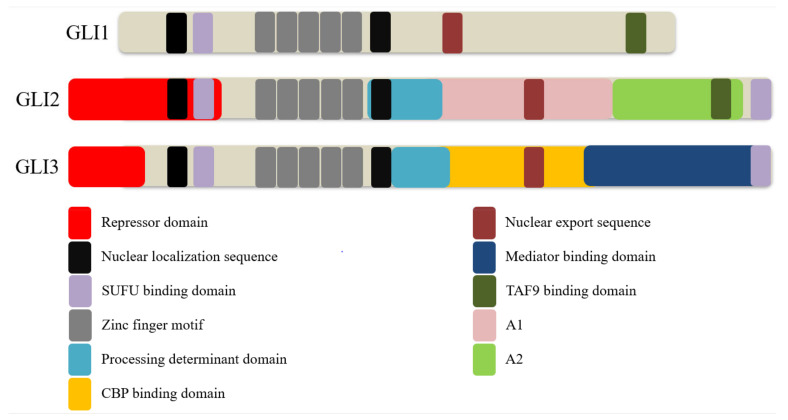
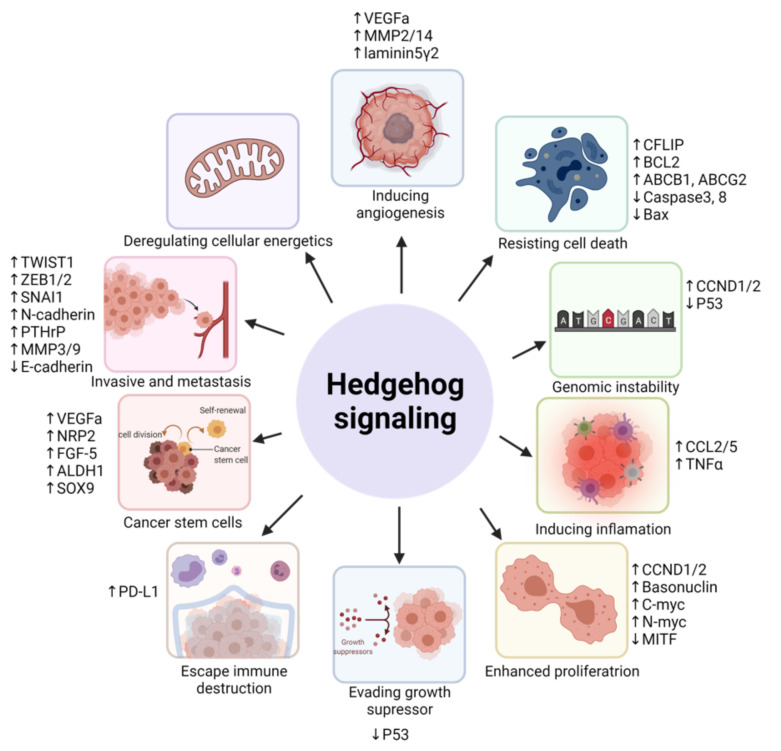
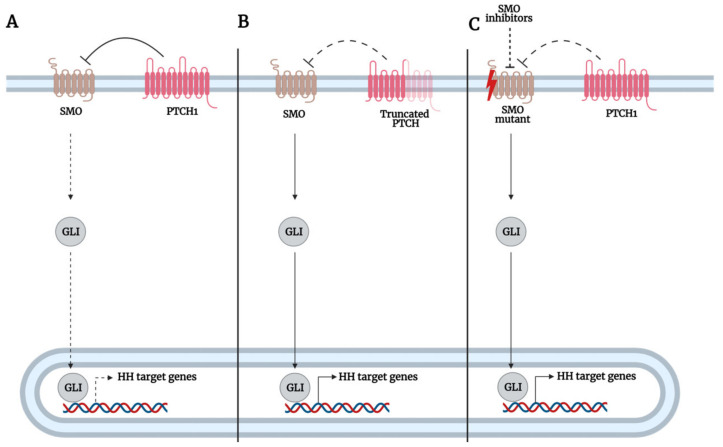
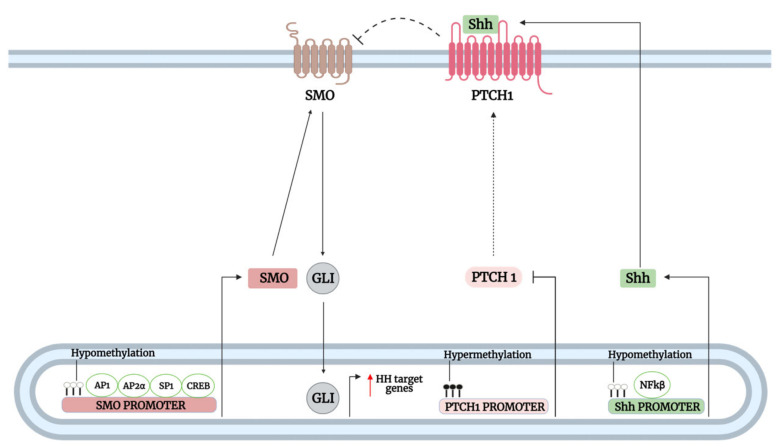
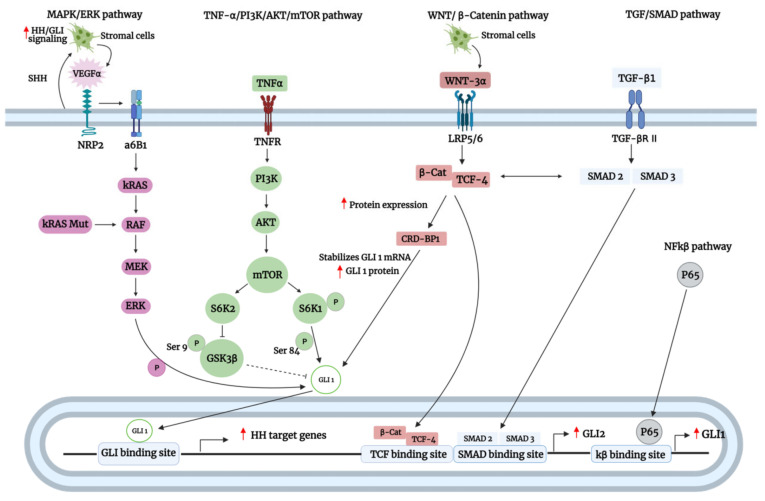
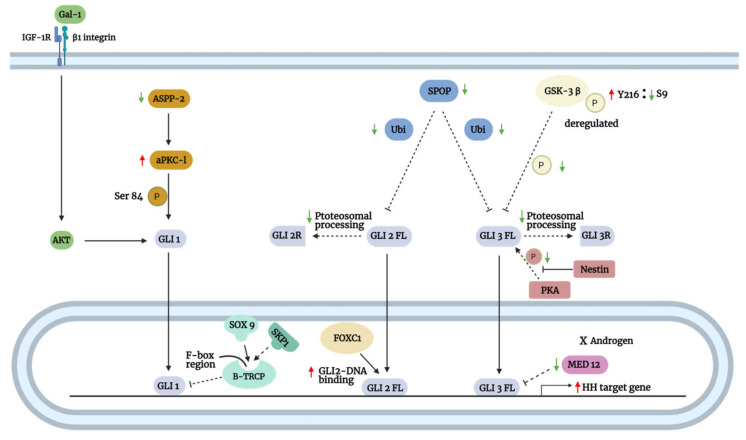
The Hedgehog (Hh)-glioma-associated oncogene homolog (GLI) signaling pathway is highly conserved among mammals, with crucial roles in regulating embryonic development as well as in cancer initiation and progression. The GLI transcription factors (GLI1, GLI2, and GLI3) are effectors of the Hh pathway and are regulated via Smoothened (SMO)-dependent and SMO-independent mechanisms. The SMO-dependent route involves the common Hh-PTCH-SMO axis, and mutations or transcriptional and epigenetic dysregulation at these levels lead to the constitutive activation of GLI transcription factors. Conversely, the SMO-independent route involves the SMO bypass regulation of GLI transcription factors by external signaling pathways and their interacting proteins or by epigenetic and transcriptional regulation of GLI transcription factors expression. Both routes of GLI activation, when dysregulated, have been heavily implicated in tumorigenesis of many known cancers, making them important targets for cancer treatment. Hence, this review describes the various SMO-dependent and SMO-independent routes of GLI regulation in the tumorigenesis of multiple cancers in order to provide a holistic view of the paradigms of hedgehog signaling networks involving GLI regulation. An in-depth understanding of the complex interplay between GLI and various signaling elements could help inspire new therapeutic breakthroughs for the treatment of Hh-GLI-dependent cancers in the future. Lastly, we have presented an up-to-date summary of the latest findings concerning the use of Hh inhibitors in clinical developmental studies and discussed the challenges, perspectives, and possible directions regarding the use of SMO/GLI inhibitors in clinical settings.
Keywords: GLI1 protein; cancer; clinical trial; epigenetic regulation; glioma-associated oncogene; hedgehog inhibitors; hedgehog pathway; mutations; noncanonical.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
