

Cellular Plasticity: A Route to Senescence Exit and Tumorigenesis
- PMID: 34572787
- PMCID: PMC8468602
- DOI: 10.3390/cancers13184561
Cellular Plasticity: A Route to Senescence Exit and Tumorigenesis
Abstract
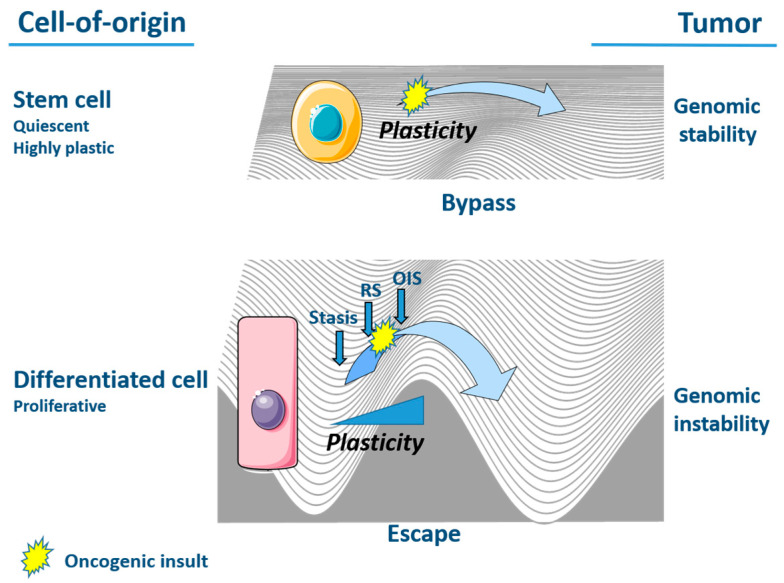
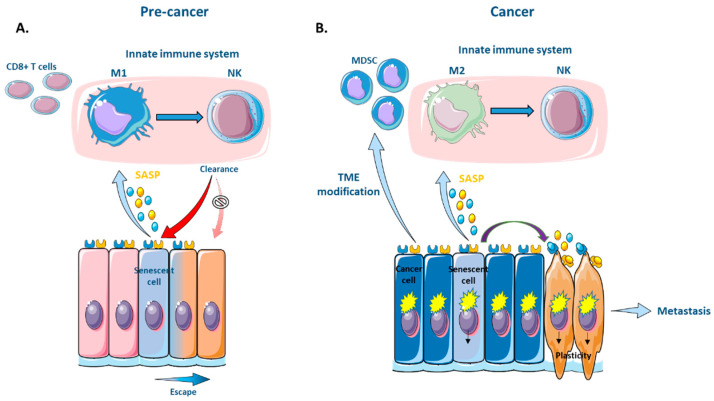
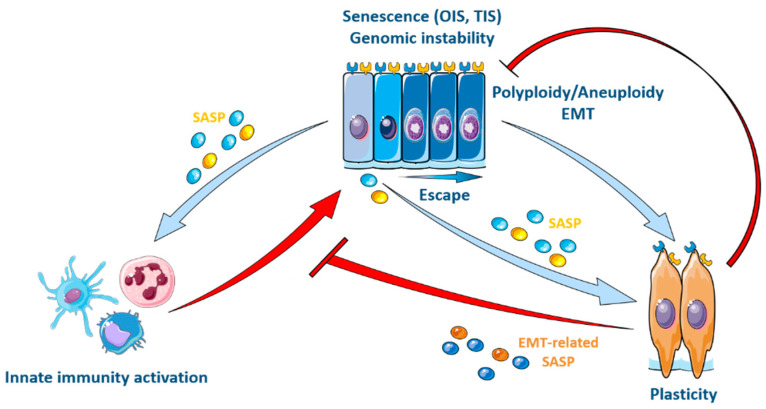
Senescence is a dynamic, multistep program that results in permanent cell cycle arrest and is triggered by developmental or environmental, oncogenic or therapy-induced stress signals. Senescence is considered as a tumor suppressor mechanism that prevents the risk of neoplastic transformation by restricting the proliferation of damaged cells. Cells undergoing senescence sustain important morphological changes, chromatin remodeling and metabolic reprogramming, and secrete pro-inflammatory factors termed senescence-associated secretory phenotype (SASP). SASP activation is required for the clearance of senescent cells by innate immunity. Therefore, escape from senescence and the associated immune editing would be a prerequisite for tumor initiation and progression as well as therapeutic resistance. One of the possible mechanisms for overcoming senescence could be the acquisition of cellular plasticity resulting from the accumulation of genomic alterations and genetic and epigenetic reprogramming. The modified composition of the SASP produced by these reprogrammed cancer cells would create a permissive environment, allowing their immune evasion. Additionally, the SASP produced by cancer cells could enhance the cellular plasticity of neighboring cells, thus hindering their recognition by the immune system. Here, we propose a comprehensive review of the literature, highlighting the role of cellular plasticity in the pro-tumoral activity of senescence in normal cells and in the cancer context.
Keywords: cellular plasticity; epithelial-mesenchymal transition; immune evasion; reprogramming; senescence.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Demaria M., Ohtani N., Youssef S.A., Rodier F., Toussaint W., Mitchell J.R., Laberge R.-M., Vijg J., Van Steeg H., Dollé M.E.T., et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell. 2014;31:722–733. doi: 10.1016/j.devcel.2014.11.012. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
