

Wild-Type KRAS Allele Effects on Druggable Targets in KRAS Mutant Lung Adenocarcinomas
- PMID: 34573384
- PMCID: PMC8467269
- DOI: 10.3390/genes12091402
Wild-Type KRAS Allele Effects on Druggable Targets in KRAS Mutant Lung Adenocarcinomas
Abstract
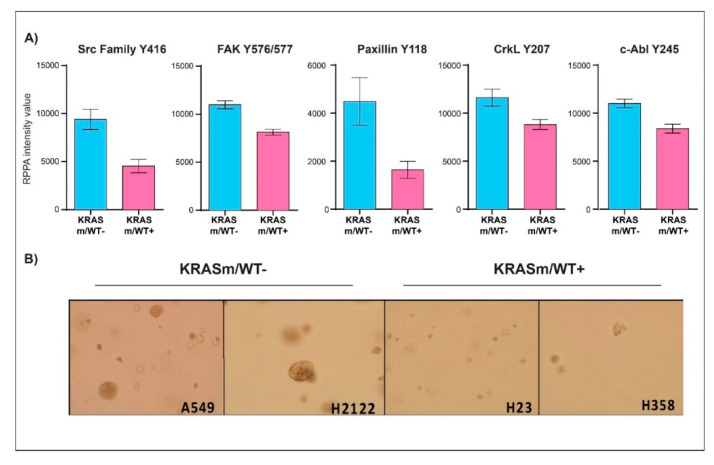
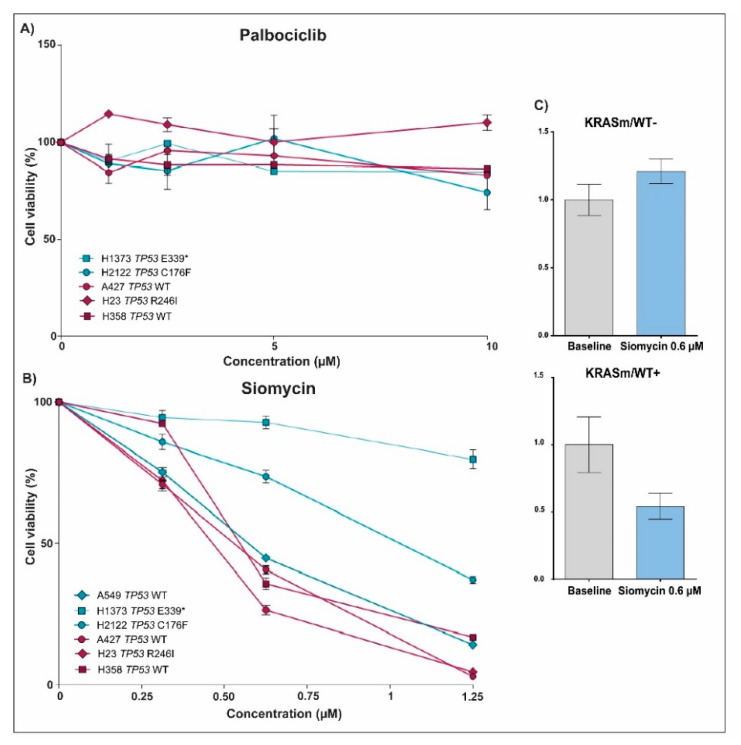
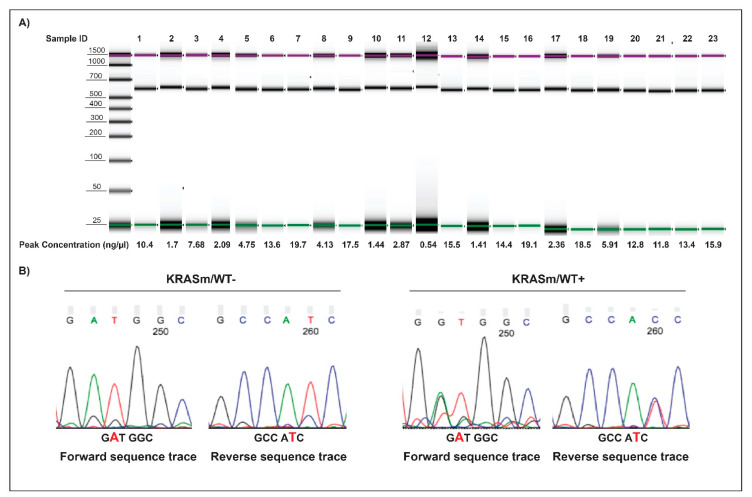
KRAS mutations are one of the most common oncogenic drivers in non-small cell lung cancer (NSCLC) and in lung adenocarcinomas in particular. Development of therapeutics targeting KRAS has been incredibly challenging, prompting indirect inhibition of downstream targets such as MEK and ERK. Such inhibitors, unfortunately, come with limited clinical efficacy, and therefore the demand for developing novel therapeutic strategies remains an urgent need for these patients. Exploring the influence of wild-type (WT) KRAS on druggable targets can uncover new vulnerabilities for the treatment of KRAS mutant lung adenocarcinomas. Using commercially available KRAS mutant lung adenocarcinoma cell lines, we explored the influence of WT KRAS on signaling networks and druggable targets. Expression and/or activation of 183 signaling proteins, most of which are targets of FDA-approved drugs, were captured by reverse-phase protein microarray (RPPA). Selected findings were validated on a cohort of 23 surgical biospecimens using the RPPA. Kinase-driven signatures associated with the presence of the KRAS WT allele were detected along the MAPK and AKT/mTOR signaling pathway and alterations of cell cycle regulators. FoxM1 emerged as a potential vulnerability of tumors retaining the KRAS WT allele both in cell lines and in the clinical samples. Our findings suggest that loss of WT KRAS impacts on signaling events and druggable targets in KRAS mutant lung adenocarcinomas.
Keywords: KRAS; drug target; non-small cell lung cancer; reverse-phase protein microarray; zygosity.
Conflict of interest statement
The authors are inventors on US Government and University assigned patents and patent applications that cover aspects of the technologies discussed such as the reverse-phase protein microarrays. As inventors, they are entitled to receive royalties as provided by US Law and George Mason University policy. M.P and E.F.P receive royalties from Theralink Technologies, Inc. E.F.P is a shareholder and consultant of Theralink Technologies and Perthera, Inc.; M.P is a consultant of Theralink Technologies, Inc.
Figures
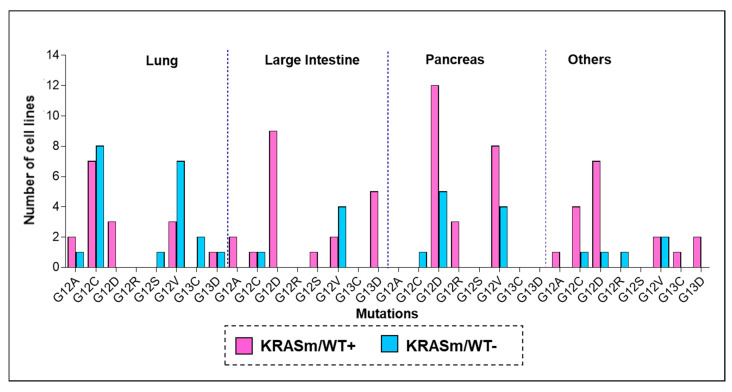
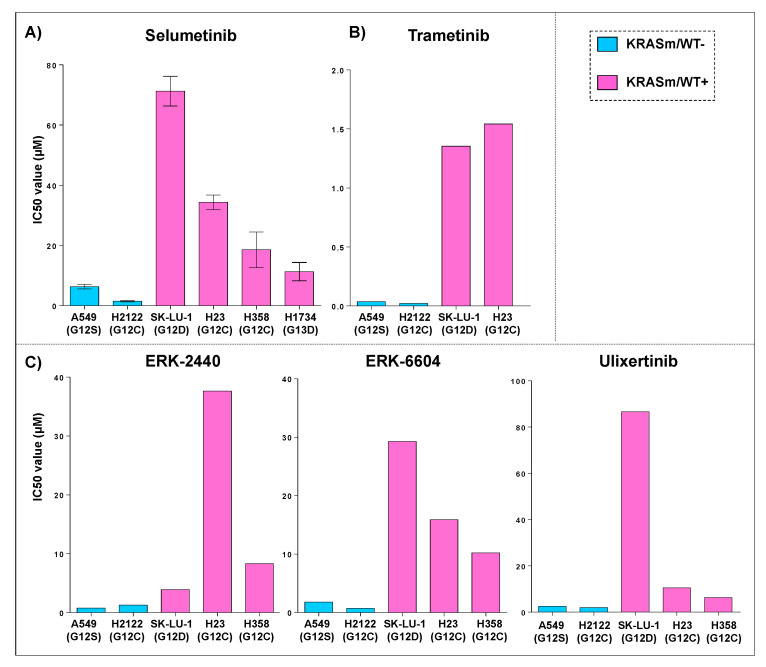
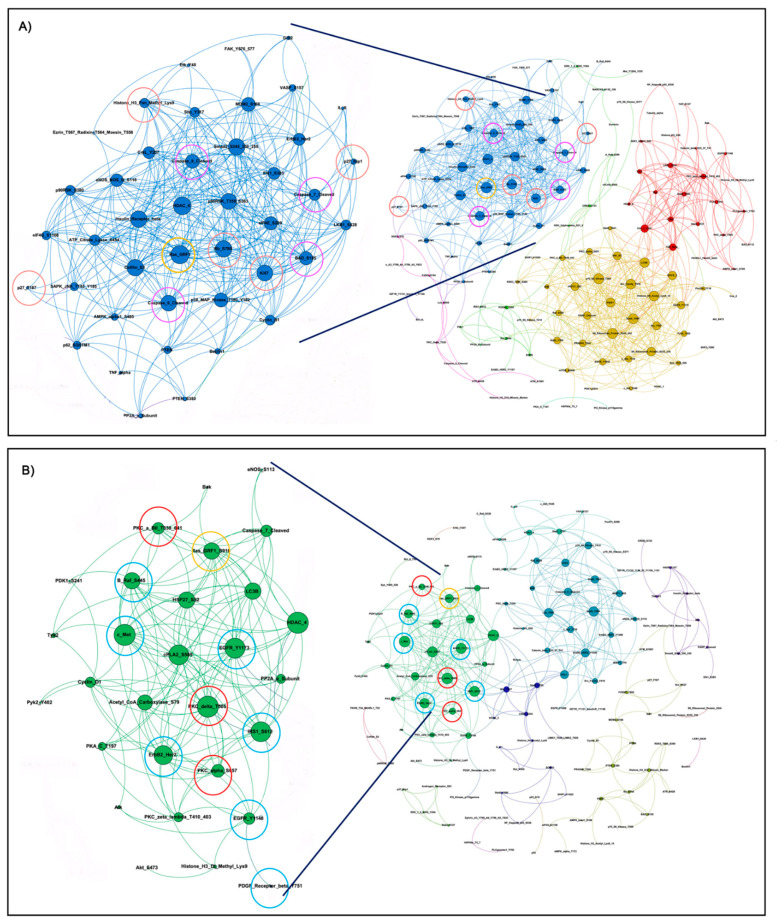
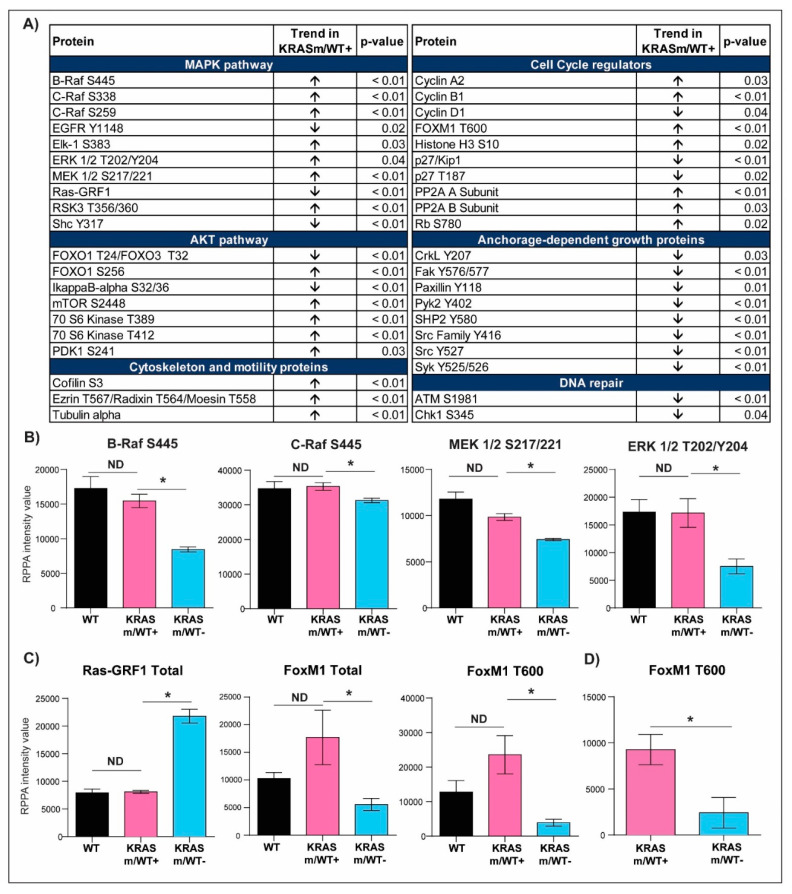
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
