

Naphthazarin Derivatives in the Light of Intra- and Intermolecular Forces
- PMID: 34577113
- PMCID: PMC8468954
- DOI: 10.3390/molecules26185642
Naphthazarin Derivatives in the Light of Intra- and Intermolecular Forces
Abstract
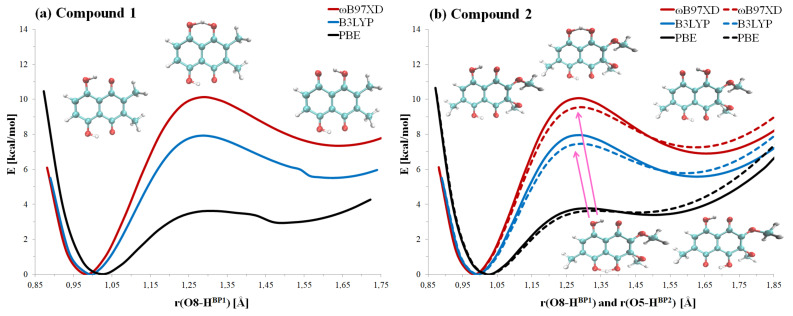
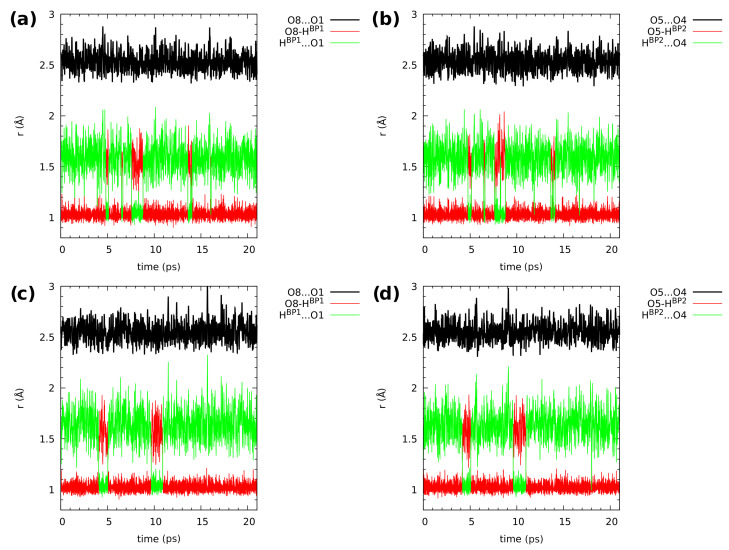
Our long-term investigations have been devoted the characterization of intramolecular hydrogen bonds in cyclic compounds. Our previous work covers naphthazarin, the parent compound of two systems discussed in the current work: 2,3-dimethylnaphthazarin (1) and 2,3-dimethoxy-6-methylnaphthazarin (2). Intramolecular hydrogen bonds and substituent effects in these compounds were analyzed on the basis of Density Functional Theory (DFT), Møller-Plesset second-order perturbation theory (MP2), Coupled Clusters with Singles and Doubles (CCSD) and Car-Parrinello Molecular Dynamics (CPMD). The simulations were carried out in the gas and crystalline phases. The nuclear quantum effects were incorporated a posteriori using the snapshots taken from ab initio trajectories. Further, they were used to solve a vibrational Schrödinger equation. The proton reaction path was studied using B3LYP, ωB97XD and PBE functionals with a 6-311++G(2d,2p) basis set. Two energy minima (deep and shallow) were found, indicating that the proton transfer phenomena could occur in the electronic ground state. Next, the electronic structure and topology were examined in the molecular and proton transferred (PT) forms. The Atoms In Molecules (AIM) theory was employed for this purpose. It was found that the hydrogen bond is stronger in the proton transferred (PT) forms. In order to estimate the dimers' stabilization and forces responsible for it, the Symmetry-Adapted Perturbation Theory (SAPT) was applied. The energy decomposition revealed that dispersion is the primary factor stabilizing the dimeric forms and crystal structure of both compounds. The CPMD results showed that the proton transfer phenomena occurred in both studied compounds, as well as in both phases. In the case of compound 2, the proton transfer events are more frequent in the solid state, indicating an influence of the environmental effects on the bridged proton dynamics. Finally, the vibrational signatures were computed for both compounds using the CPMD trajectories. The Fourier transformation of the autocorrelation function of atomic velocity was applied to obtain the power spectra. The IR spectra show very broad absorption regions between 700 cm-1-1700 cm-1 and 2300 cm-1-3400 cm-1 in the gas phase and 600 cm-1-1800 cm-1 and 2200 cm-1-3400 cm-1 in the solid state for compound 1. The absorption regions for compound 2 were found as follows: 700 cm-1-1700 cm-1 and 2300 cm-1-3300 cm-1 for the gas phase and one broad absorption region in the solid state between 700 cm-1 and 3100 cm-1. The obtained spectroscopic features confirmed a strong mobility of the bridged protons. The inclusion of nuclear quantum effects showed a stronger delocalization of the bridged protons.
Keywords: AIM; CCSD; CPMD; DFT; MP2; SAPT; crystalline phase; gas phase; intramolecular hydrogen bonds; nuclear quantum effects.
Conflict of interest statement
The authors declare no conflict of interest.
Figures








Similar articles
-
Non-Covalent Forces in Naphthazarin-Cooperativity or Competition in the Light of Theoretical Approaches.Int J Mol Sci. 2021 Jul 27;22(15):8033. doi: 10.3390/ijms22158033. Int J Mol Sci. 2021. PMID: 34360798 Free PMC article.
-
Revealing Intra- and Intermolecular Interactions Determining Physico-Chemical Features of Selected Quinolone Carboxylic Acid Derivatives.Molecules. 2022 Apr 1;27(7):2299. doi: 10.3390/molecules27072299. Molecules. 2022. PMID: 35408698 Free PMC article.
-
Competition of Intra- and Intermolecular Forces in Anthraquinone and Its Selected Derivatives.Molecules. 2021 Jun 6;26(11):3448. doi: 10.3390/molecules26113448. Molecules. 2021. PMID: 34204133 Free PMC article.
-
Sensitivity of Intra- and Intermolecular Interactions of Benzo[h]quinoline from Car-Parrinello Molecular Dynamics and Electronic Structure Inspection.Int J Mol Sci. 2021 May 14;22(10):5220. doi: 10.3390/ijms22105220. Int J Mol Sci. 2021. PMID: 34069244 Free PMC article.
-
Mini Review: Quantum Confinement of Atomic and Molecular Clusters in Carbon Nanotubes.Front Chem. 2021 Dec 8;9:796890. doi: 10.3389/fchem.2021.796890. eCollection 2021. Front Chem. 2021. PMID: 34957050 Free PMC article. Review.
Cited by
-
Interrogation of the Intermolecular Forces That Drive Bulk Properties of Molecular Crystals with Terahertz Spectroscopy and Density Functional Theory.Cryst Growth Des. 2025 May 23;25(11):3697-3706. doi: 10.1021/acs.cgd.5c00007. eCollection 2025 Jun 4. Cryst Growth Des. 2025. PMID: 40491562 Free PMC article.
-
Exploring Intra- and Intermolecular Interactions in Selected N-Oxides-The Role of Hydrogen Bonds.Molecules. 2022 Jan 25;27(3):792. doi: 10.3390/molecules27030792. Molecules. 2022. PMID: 35164056 Free PMC article.
-
Comprehensive Empirical Model of Substitution-Influence on Hydrogen Bonding in Aromatic Schiff Bases.Int J Mol Sci. 2022 Oct 18;23(20):12439. doi: 10.3390/ijms232012439. Int J Mol Sci. 2022. PMID: 36293296 Free PMC article.
-
Intramolecular Hydrogen Bonding 2021.Molecules. 2021 Oct 19;26(20):6319. doi: 10.3390/molecules26206319. Molecules. 2021. PMID: 34684899 Free PMC article.
References
-
- Desiraju G.R., Steiner T. The Weak Hydrogen Bond. Oxford University Press; Oxford, UK: 2001. Other weak and non-conventional hydrogen bonds; pp. 122–292. - DOI
-
- Honacker C., Kappelt B., Jabłoński M., Hepp A., Layh M., Rogel F., Uhl W. Aluminium Functionalized Germanes: Intramolecular Activation of Ge-H Bonds, Formation of a Dihydrogen Bond and Facile Hydrogermylation of Unsaturated Substrates. Eur. J. Inorg. Chem. 2019;2019:3287–3300. doi: 10.1002/ejic.201900543. - DOI
-
- Hobza P., Zahradník R., Müller-Dethlefs K. The World of Non-Covalent Interactions: 2006. Collect. Czechoslov. Chem. Commun. 2006;71:443–531. doi: 10.1135/cccc20060443. - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
