

Structure-Guided Creation of an Anti-HA Stalk Antibody F11 Derivative That Neutralizes Both F11-Sensitive and -Resistant Influenza A(H1N1)pdm09 Viruses
- PMID: 34578314
- PMCID: PMC8473006
- DOI: 10.3390/v13091733
Structure-Guided Creation of an Anti-HA Stalk Antibody F11 Derivative That Neutralizes Both F11-Sensitive and -Resistant Influenza A(H1N1)pdm09 Viruses
Abstract
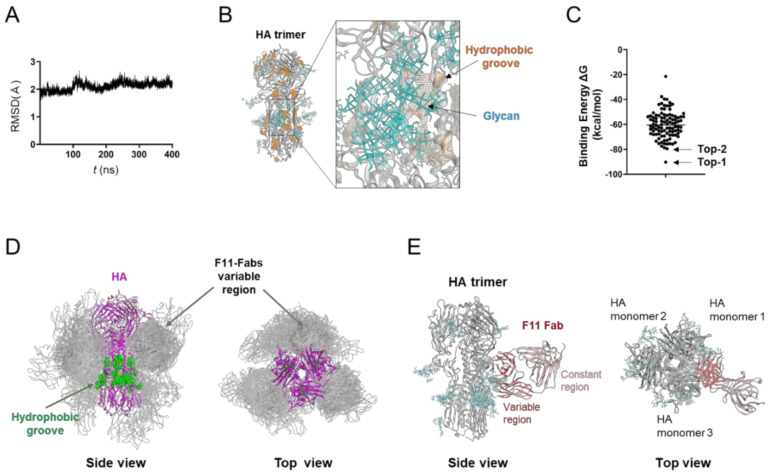
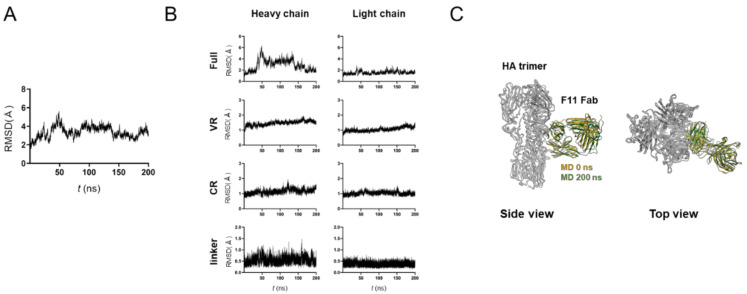
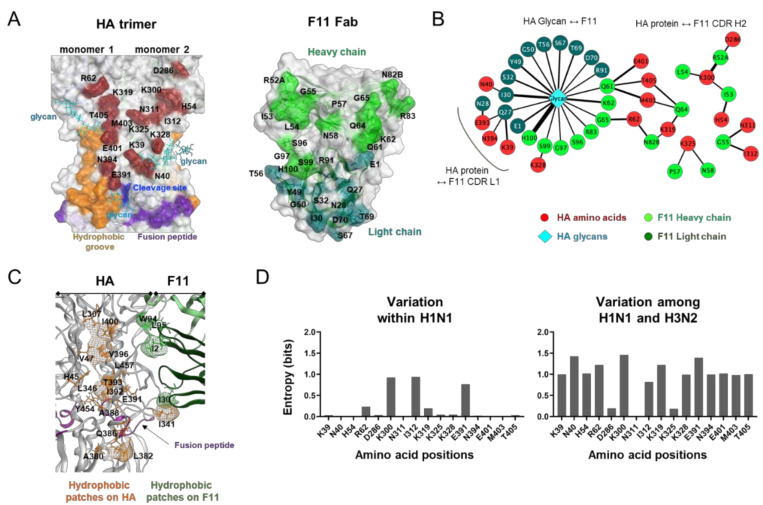
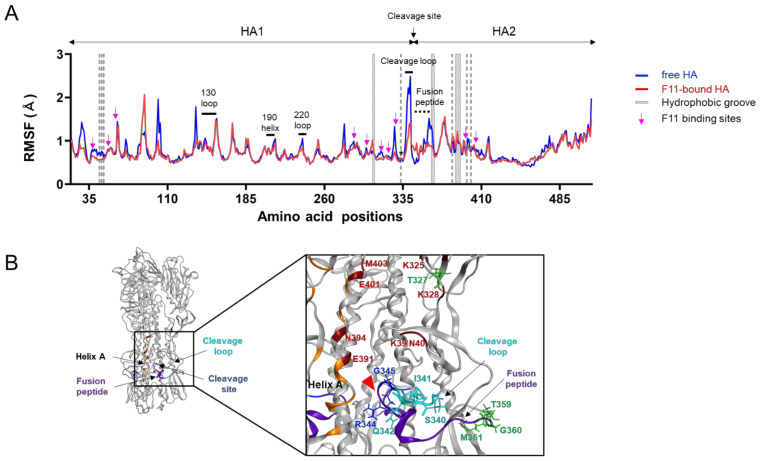
The stalk domain of influenza virus envelope glycoprotein hemagglutinin (HA) constitutes the axis connecting the head and transmembrane domains, and plays pivotal roles in conformational rearrangements of HA for virus infection. Here we characterized molecular interactions between the anti-HA stalk neutralization antibody F11 and influenza A(H1N1)pdm09 HA to understand the structural basis of the actions and modifications of this antibody. In silico structural analyses using a model of the trimeric HA ectodomain indicated that the F11 Fab fragment has physicochemical properties, allowing it to crosslink two HA monomers by binding to a region near the proteolytic cleavage site of the stalk domain. Interestingly, the F11 binding allosterically caused a marked suppression of the structural dynamics of the HA cleavage loop and flanking regions. Structure-guided mutagenesis of the F11 antibody revealed a critical residue in the F11 light chain for the F11-mediated neutralization. Finally, the mutagenesis led to identification of a unique F11 derivative that can neutralize both F11-sensitive and F11-resistant A(H1N1)pdm09 viruses. These results raise the possibility that F11 sterically and physically disturbs proteolytic cleavage of HA for the ordered conformational rearrangements and suggest that in silico guiding experiments can be useful to create anti-HA stalk antibodies with new phenotypes.
Keywords: anti-HA stalk antibody; antibody modification; influenza virus; molecular dynamics simulation; molecular interactions; neutralization assay.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures
Similar articles
-
Potential Role of Nonneutralizing IgA Antibodies in Cross-Protective Immunity against Influenza A Viruses of Multiple Hemagglutinin Subtypes.J Virol. 2020 Jun 1;94(12):e00408-20. doi: 10.1128/JVI.00408-20. Print 2020 Jun 1. J Virol. 2020. PMID: 32269119 Free PMC article.
-
Extending the Stalk Enhances Immunogenicity of the Influenza Virus Neuraminidase.J Virol. 2019 Aug 28;93(18):e00840-19. doi: 10.1128/JVI.00840-19. Print 2019 Sep 15. J Virol. 2019. PMID: 31375573 Free PMC article.
-
The influenza virus hemagglutinin head evolves faster than the stalk domain.Sci Rep. 2018 Jul 11;8(1):10432. doi: 10.1038/s41598-018-28706-1. Sci Rep. 2018. PMID: 29992986 Free PMC article.
-
Human Monoclonal Antibody 81.39a Effectively Neutralizes Emerging Influenza A Viruses of Group 1 and 2 Hemagglutinins.J Virol. 2016 Nov 14;90(23):10446-10458. doi: 10.1128/JVI.01284-16. Print 2016 Dec 1. J Virol. 2016. PMID: 27630240 Free PMC article.
-
Structural studies on antibody recognition and neutralization of viruses.Curr Opin Virol. 2011 Aug;1(2):150-6. doi: 10.1016/j.coviro.2011.05.020. Curr Opin Virol. 2011. PMID: 21887208 Free PMC article. Review.
Cited by
-
Cis-Allosteric Regulation of HIV-1 Reverse Transcriptase by Integrase.Viruses. 2022 Dec 21;15(1):31. doi: 10.3390/v15010031. Viruses. 2022. PMID: 36680070 Free PMC article.
-
Loxapine inhibits replication of hepatitis A virus in vitro and in vivo by targeting viral protein 2C.PLoS Pathog. 2024 Mar 13;20(3):e1012091. doi: 10.1371/journal.ppat.1012091. eCollection 2024 Mar. PLoS Pathog. 2024. PMID: 38478584 Free PMC article.
-
Unique Mode of Antiviral Action of a Marine Alkaloid against Ebola Virus and SARS-CoV-2.Viruses. 2022 Apr 15;14(4):816. doi: 10.3390/v14040816. Viruses. 2022. PMID: 35458549 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
