

High prevalence of multilocus pathogenic variation in neurodevelopmental disorders in the Turkish population
- PMID: 34582790
- PMCID: PMC8546040
- DOI: 10.1016/j.ajhg.2021.08.009
High prevalence of multilocus pathogenic variation in neurodevelopmental disorders in the Turkish population
Abstract
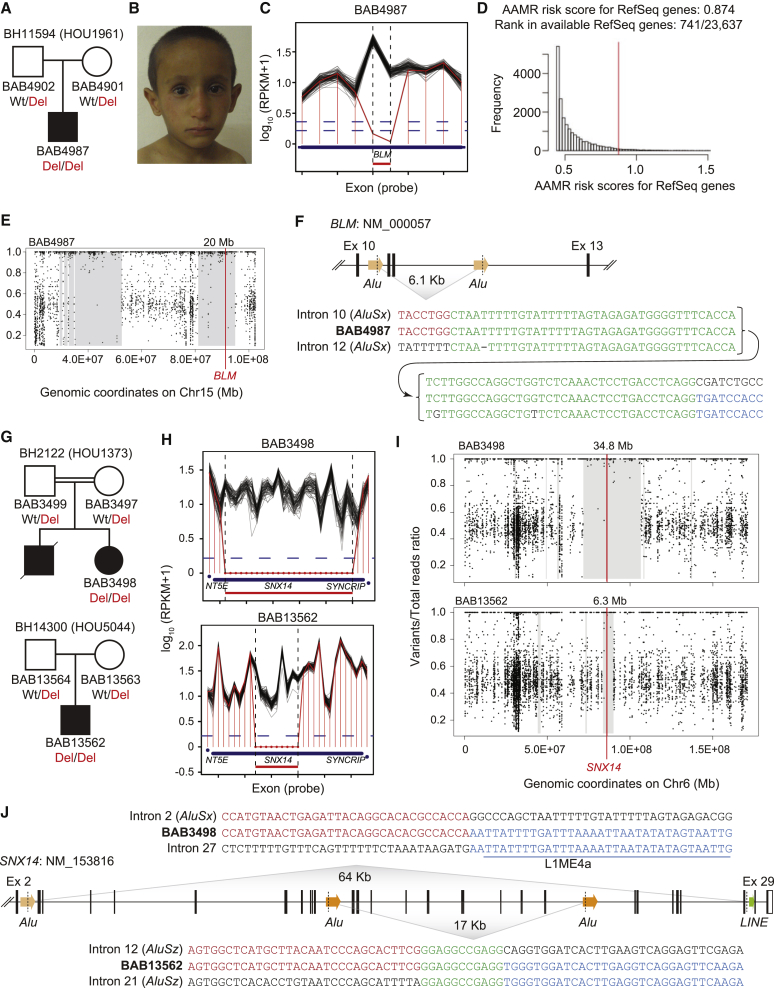
Neurodevelopmental disorders (NDDs) are clinically and genetically heterogenous; many such disorders are secondary to perturbation in brain development and/or function. The prevalence of NDDs is > 3%, resulting in significant sociocultural and economic challenges to society. With recent advances in family-based genomics, rare-variant analyses, and further exploration of the Clan Genomics hypothesis, there has been a logarithmic explosion in neurogenetic "disease-associated genes" molecular etiology and biology of NDDs; however, the majority of NDDs remain molecularly undiagnosed. We applied genome-wide screening technologies, including exome sequencing (ES) and whole-genome sequencing (WGS), to identify the molecular etiology of 234 newly enrolled subjects and 20 previously unsolved Turkish NDD families. In 176 of the 234 studied families (75.2%), a plausible and genetically parsimonious molecular etiology was identified. Out of 176 solved families, deleterious variants were identified in 218 distinct genes, further documenting the enormous genetic heterogeneity and diverse perturbations in human biology underlying NDDs. We propose 86 candidate disease-trait-associated genes for an NDD phenotype. Importantly, on the basis of objective and internally established variant prioritization criteria, we identified 51 families (51/176 = 28.9%) with multilocus pathogenic variation (MPV), mostly driven by runs of homozygosity (ROHs) - reflecting genomic segments/haplotypes that are identical-by-descent. Furthermore, with the use of additional bioinformatic tools and expansion of ES to additional family members, we established a molecular diagnosis in 5 out of 20 families (25%) who remained undiagnosed in our previously studied NDD cohort emanating from Turkey.
Keywords: Alu-Alu mediated rearrangement (AAMR); exome reanalysis; identity-by-descent (IBD); multilocus pathogenic variation; neurodevelopmental disorders; runs of homozygosity (ROH); whole-genome sequencing.
Copyright © 2021 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests J.R.L. has stock ownership in 23andMe, is a paid consultant for the Regeneron Genetics Center, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics (BG) Laboratories. J.R.L. serves on the Scientific Advisory Board of BG. Other authors have no potential conflicts to report.
Figures






References
-
- Parenti I., Rabaneda L.G., Schoen H., Novarino G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci. 2020;43:608–621. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
