

Enhanced Heart Failure in Redox-Dead Cys17Ser PKARIα Knock-In Mice
- PMID: 34583520
- PMCID: PMC8649132
- DOI: 10.1161/JAHA.121.021985
Enhanced Heart Failure in Redox-Dead Cys17Ser PKARIα Knock-In Mice
Abstract
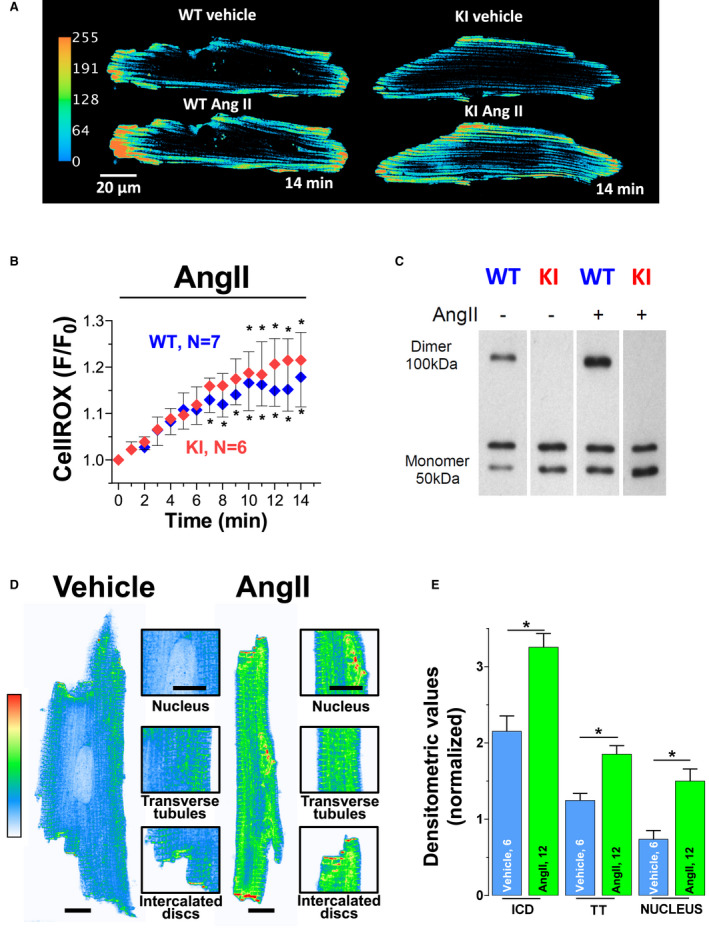
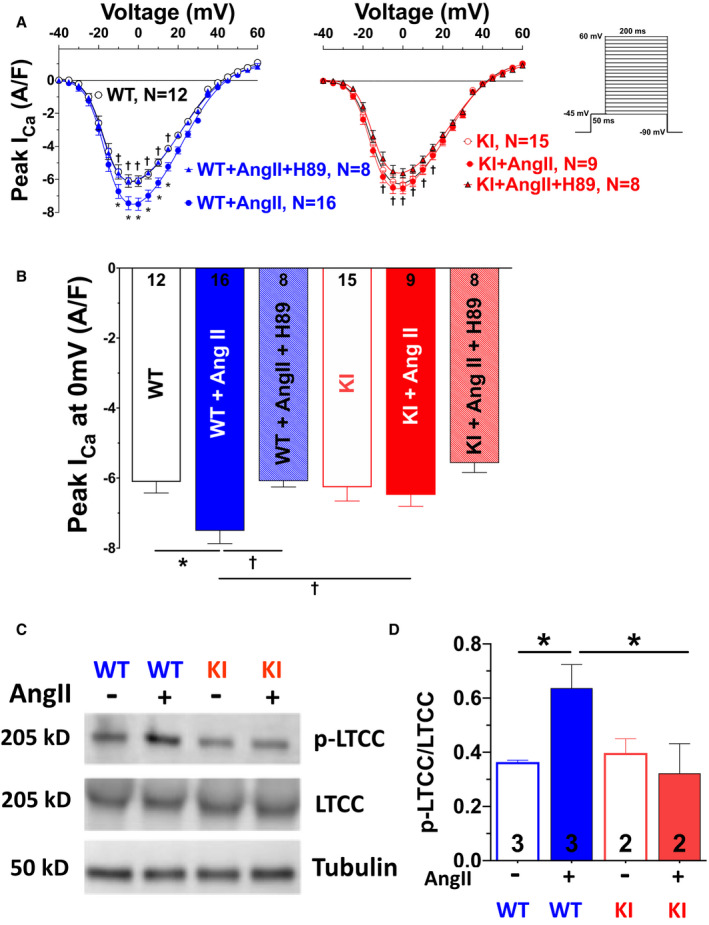
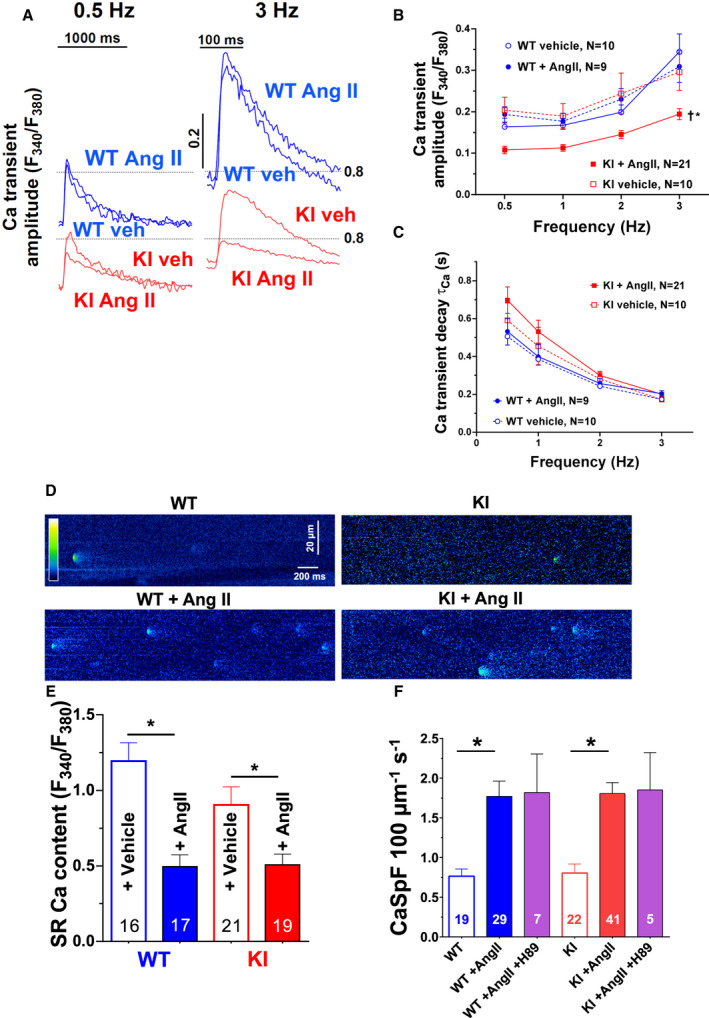
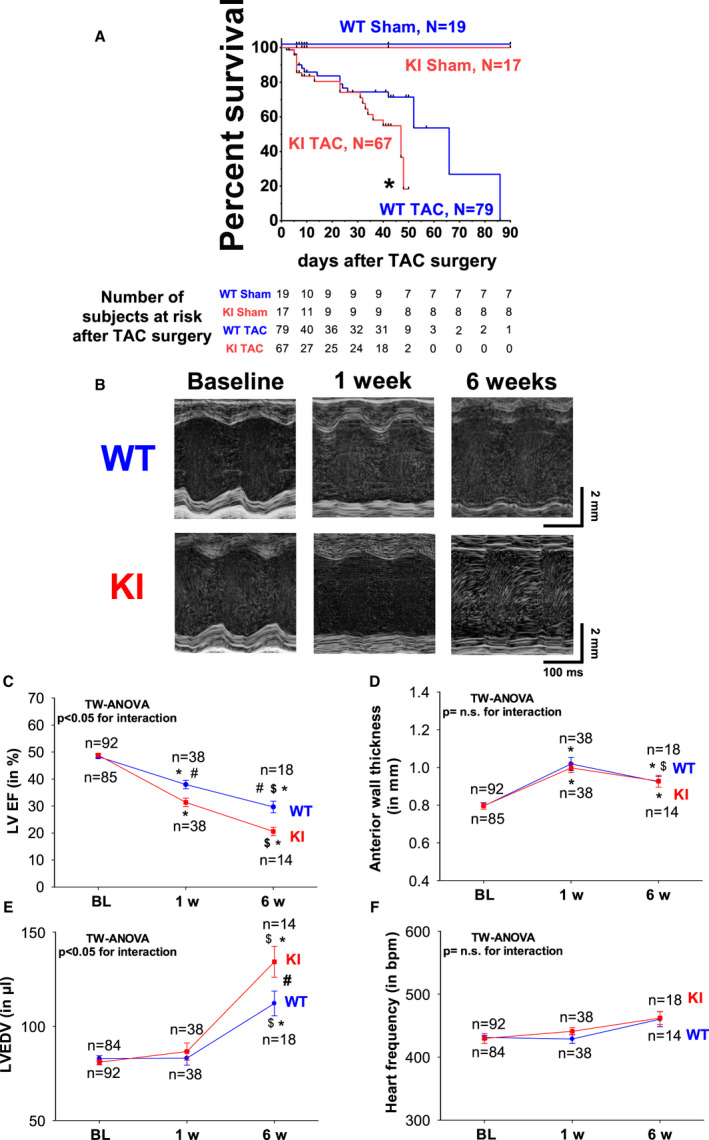
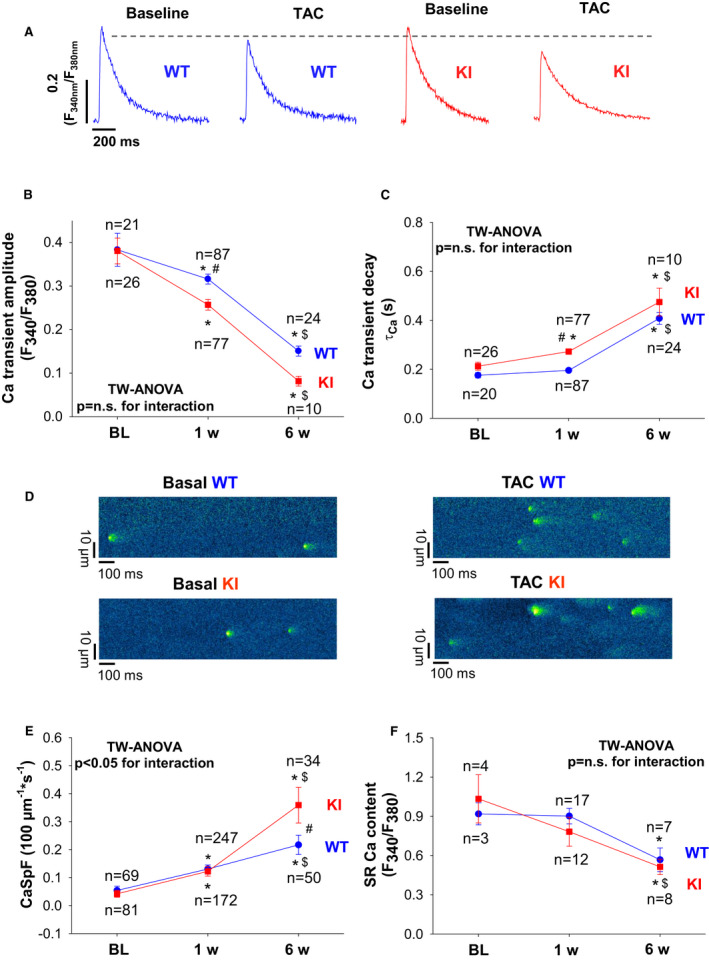
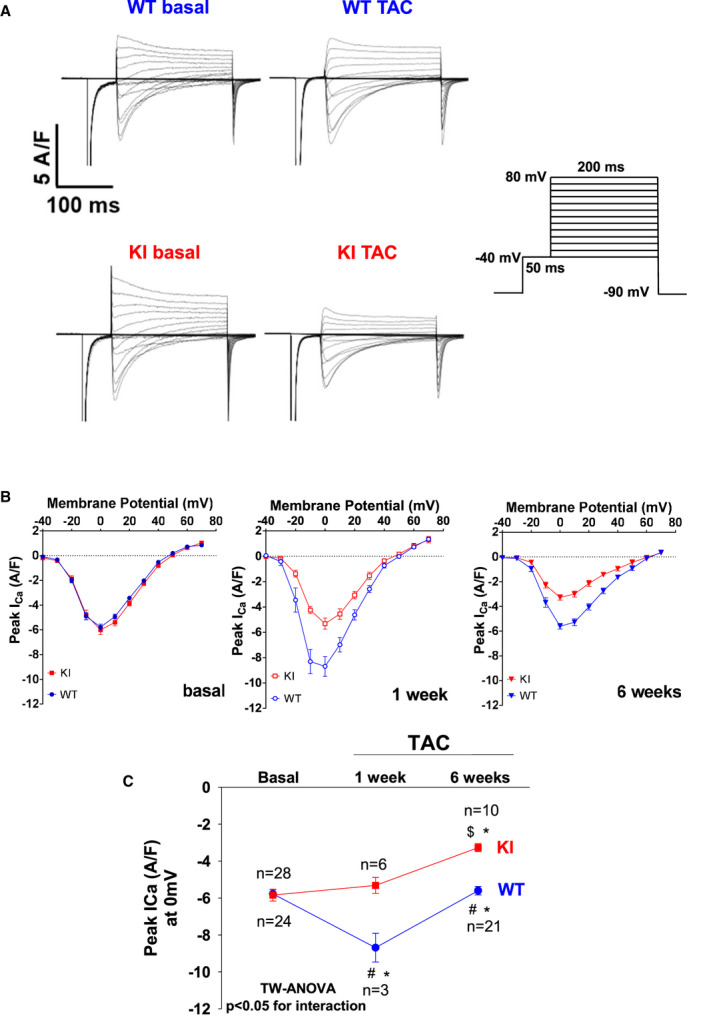
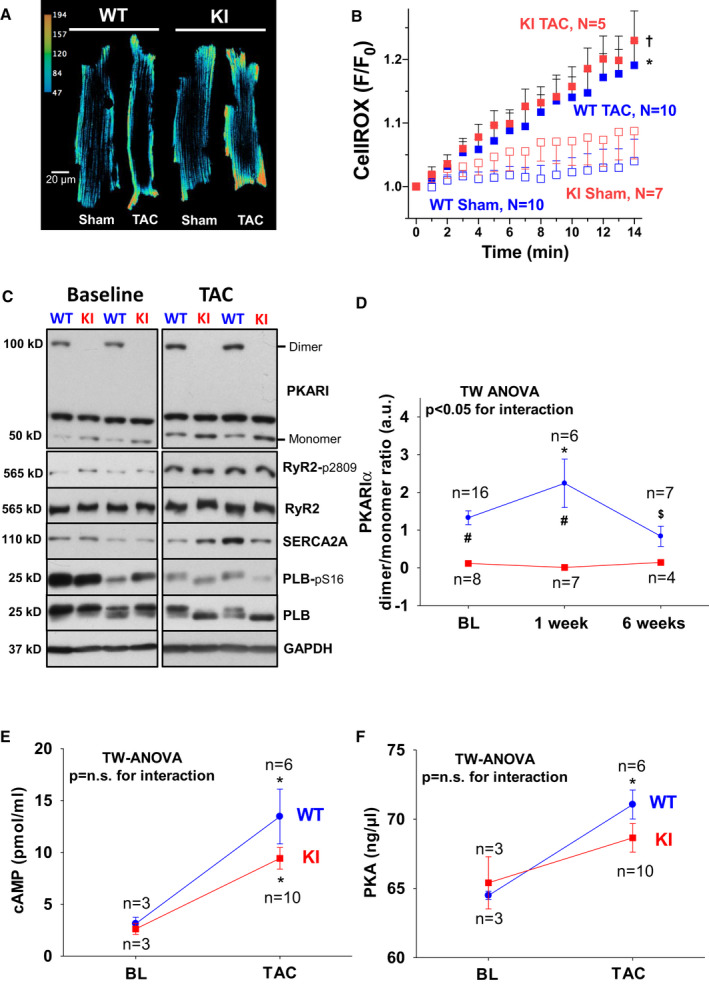
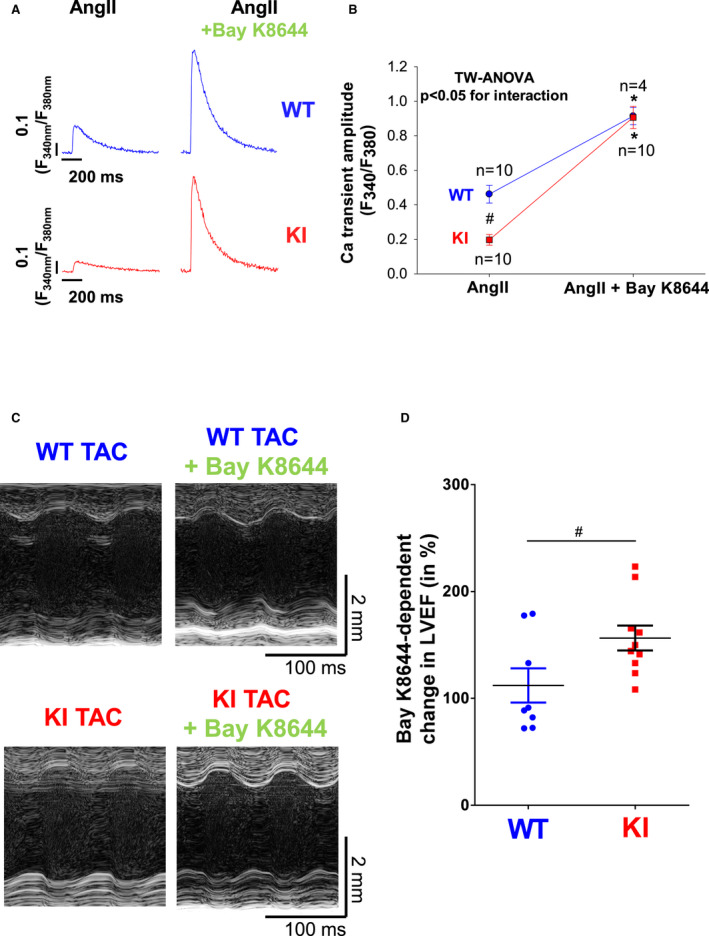
Background PKARIα (protein kinase A type I-α regulatory subunit) is redox-active independent of its physiologic agonist cAMP. However, it is unknown whether this alternative mechanism of PKARIα activation may be of relevance to cardiac excitation-contraction coupling. Methods and Results We used a redox-dead transgenic mouse model with homozygous knock-in replacement of redox-sensitive cysteine 17 with serine within the regulatory subunits of PKARIα (KI). Reactive oxygen species were acutely evoked by exposure of isolated cardiac myocytes to AngII (angiotensin II, 1 µmol/L). The long-term relevance of oxidized PKARIα was investigated in KI mice and their wild-type (WT) littermates following transverse aortic constriction (TAC). AngII increased reactive oxygen species in both groups but with RIα dimer formation in WT only. AngII induced translocation of PKARI to the cell membrane and resulted in protein kinase A-dependent stimulation of ICa (L-type Ca current) in WT with no effect in KI myocytes. Consequently, Ca transients were reduced in KI myocytes as compared with WT cells following acute AngII exposure. Transverse aortic constriction-related reactive oxygen species formation resulted in RIα oxidation in WT but not in KI mice. Within 6 weeks after TAC, KI mice showed an enhanced deterioration of contractile function and impaired survival compared with WT. In accordance, compared with WT, ventricular myocytes from failing KI mice displayed significantly reduced Ca transient amplitudes and lack of ICa stimulation. Conversely, direct pharmacological stimulation of ICa using Bay K8644 rescued Ca transients in AngII-treated KI myocytes and contractile function in failing KI mice in vivo. Conclusions Oxidative activation of PKARIα with subsequent stimulation of ICa preserves cardiac function in the setting of acute and chronic oxidative stress.
Keywords: heart failure; pressure overload; protein kinase A; redox.
Conflict of interest statement
None.
Figures
References
-
- Gill JS, McKenna WJ, Camm AJ. Free radicals irreversibly decrease Ca2+ currents in isolated guinea‐pig ventricular myocytes. Eur J Pharmacol. 1995;292:337–340. - PubMed
-
- Morris TE, Sulakhe PV. Sarcoplasmic reticulum Ca(2+)‐pump dysfunction in rat cardiomyocytes briefly exposed to hydroxyl radicals. Free Radic Biol Med. 1997;22:37–47. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- RG/17/16/33294/BHF_/British Heart Foundation/United Kingdom
- MR/R01065X/1/MRC_/Medical Research Council/United Kingdom
- MR/R01065X/2/MRC_/Medical Research Council/United Kingdom
- RG/20/3/34823/BHF_/British Heart Foundation/United Kingdom
- G0700320/MRC_/Medical Research Council/United Kingdom
- MR/P023150/2/MRC_/Medical Research Council/United Kingdom
- PG/17/39/33027/BHF_/British Heart Foundation/United Kingdom
- PG/19/33/34385/BHF_/British Heart Foundation/United Kingdom
- RG/13/11/30384/BHF_/British Heart Foundation/United Kingdom
- CH/1999001/11735/BHF_/British Heart Foundation/United Kingdom
- MR/P023150/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
